Home / Education / Tutorials / EMBOSS -> JEMBOSS User manual -> 序列比對
|
序列比對分析 Sequence Alignment
|
|||||||
|
|
|||||||
|
|||||||
|
|||||||
|
|||||||
|
一、雙序列並列分析:dottup, dotmatcher, needle, water |
|||||||
| 比對兩個序列間相似性,可以先以Dotplot觀察相似的區域,再以Needle或water 程式進行比對,得知兩條序列相似度。Dotplot 分析是以點陣圖來觀察兩條序列整體的 相似的情形,有時也可用來比對 cDNA 和genomic sequence,作為判斷 exon 位置的參考 ,EMBOSS中可選擇Dottup或Dotmatcher兩個程式進行Dotplot分析。至於兩條序列相似度 的比對可使用needle或water 程式,needle是用來尋找兩條序列整體的最佳排列方式之用 (global alignment); 而water則是用來尋找兩段序列間最佳排列區域(local alignment) 。needle是將兩條序列從頭到尾完全並列的分析,常會在序列中間插入許多的gap;water 則只列出最相似的區域兩條序列,其餘略去不列出來。以下就這四個程式一一作介紹。 | |||||||
| Dottup for Matched words | |||||||
| Dottup 程式執行的方式是在給定序列長度下 (word size) 逐一比對,找到完全相同的序列片 段 (即:word size 為10,且有10個連續的 residues 完全相同)。其比對步驟為給定兩條序列,設word=10,程式會依 序比對兩條序列的第 1-10,2-11,3-12,一段一段交互比,只要在相對位置有相同的 residue 就以一個 "點" (dotplot) 來表示,找到兩段序列有 10 個 residues完全相同的片段,就會在結果中顯示出來。 | |||||||
|
|
|||||||
 |
|||||||
| Dottup 操作步驟: | |||||||
|
|
進入 dottup | ||||||
|
|
輸入序列,有 3種方法可供選擇: | ||||||
|
1.
|
由 file /database:鍵入database:accession number。 | ||||||
|
2.
|
copy /paste:直接貼上序列。 | ||||||
|
3.
|
擷取PC中的序列檔案:點選"List of files", 再將您電腦中適當的序列檔案輸入即可。 | ||||||
|
|
以下列二條序列為例,選定由 file/database 輸入 輸入序列(1) embl:HSPDGA4 輸入序列(2) embl:HSA238420 |
||||||
|
|
在 required section 設 word size =10 | ||||||
|
|
在 out put section選擇輸出格式-- (選擇 stretch axes) | ||||||
|
1.
|
stretch axes:以 x, y 軸伸展的 dotplot 圖形顯示比對結果。 | ||||||
|
2.
|
display as data:比對圖形的文字檔 | ||||||
|
3.
|
daw a box around dotbox:以長方格圖形顯示比對結果。 | ||||||
|
|
注意:在執行區右下角可選擇 mode,"batch" (背景執行) 或是 "interactive" (在使用中視窗執行 ) | ||||||
|
|
按 Go | ||||||
|
|
結果視窗跳出後存檔,記得副檔名為 ".png" | ||||||
|
|
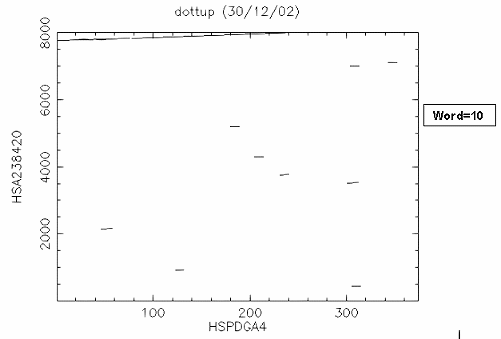
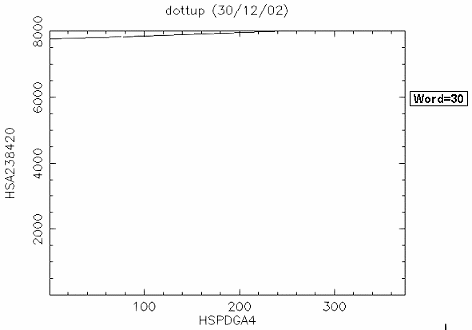
比對結果: 由於 "embl:HSPDGA4" 是 PDGF A chain exon4 的序列,而 "embl:HSA238420" 是 PDGF A chain gene exon 1~4 的序列,所以很清楚的可以看到 exon4 位於 PDGF A chain gene 約 7700~8000 的位置。如果設 window size=30 結果會更清楚。 |
||||||
 |
|||||||
 |
|||||||
| Dotmatcher for threshold similarity | |||||||
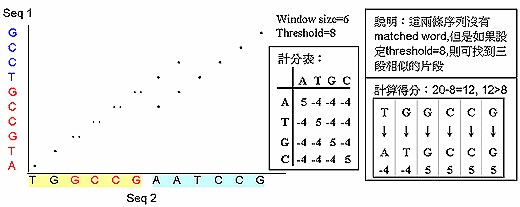
| Dotmatcher 執行的方式則是設定 threshold 的方式來圖示序列的相似度。當兩條序列在給定 的 word size 中找不到完全相同的序列 (如 word size 為10,但 10個 residues 中只有 7個 residue 相同) 時可以採用 dotmatcher 進行 dotplot 分析。其比對步驟為給定兩條序列,設 word=6,Threshold=8, 程式會依序比對兩條序列的第 1-6,2-7,3-8,一段一段交互比對,只要在相對位置有相同的 residue 就以一個 "點" 來表示,兩段序列 residues 比對得分≧8,就會在結果中顯示出來,如下圖。 | |||||||
 |
|||||||
| Dotmatcher 操作步驟: | |||||||
|
|
進入 dotmatcher | ||||||
|
|
輸入序列,有3種方法可供選擇: | ||||||
|
1.
|
由 file /database:鍵入database:accession number。 | ||||||
|
2.
|
copy /paste:直接貼上序列。 | ||||||
|
3.
|
擷取PC中的序列檔案:點選 "List of files", 再將您電腦中適當的序列檔案輸入即可。 | ||||||
|
|
以下列二條序列為例,選定由 file/database 輸入 輸入序列(1) embl:HSPDGA4 輸入序列(2) embl:HSA238420 |
||||||
|
|
選擇 Advanced options,拉下視窗,設 word size =10,threshold=50 (或threshold=23) | ||||||
|
|
選擇 out put section | ||||||
|
1.
|
stretch axes:以 x, y 軸伸展的 dotplot 圖形顯示比對結果。 | ||||||
|
2.
|
display as data:選擇 stretch axes | ||||||
|
|
注意:在執行區右下角可選擇 mode,"batch" (背景執行) 或是 "interactive" (在使用中視窗執行) | ||||||
|
|
按 GO | ||||||
|
|
結果視窗跳出後存檔,記得副檔名為 ".png" | ||||||
|
|
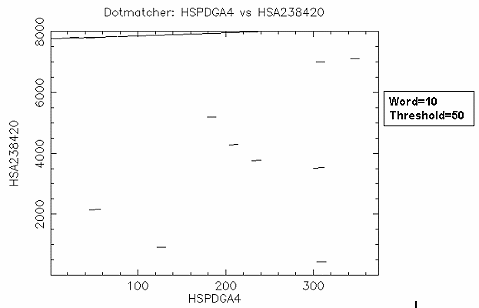
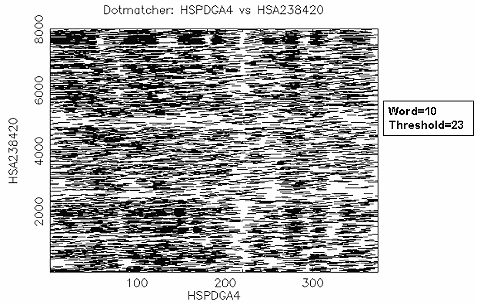
比對結果: 由於 "embl:HSPDGA4" 是 PDGF A chain exon4 的序列,而 "embl:HSA238420" 是 PDGF A chain gene exon1~4 的序列,設 threshold=50 表示 exact match, (因為DNA sequence match給5分, word=10, 每一個 residue 都 match 的分數是50分) 所以結果和 dottup 一樣,很清楚的可以看到 exon4 位於 PDGF A chain gene 約 7700~8000 的位置。如果設 threshold =23 (default 值),則整條序列相似的區域都會顯示出來, 難以區別。因此使用 dotmatcher 時,要記得設不同的threshold 值多 run 幾次,以得到您所需要的序列比對 overview。 |
||||||
 |
|||||||
 |
|||||||
| Needle:以Global Alignment進行序列的兩兩比對 | |||||||
| needle操作步驟: | |||||||
|
|
Go to : needle | ||||||
|
|
輸入序列,有3種方法可供選擇: | ||||||
|
1.
|
由 file /database:鍵入database:accession number。 | ||||||
|
2.
|
copy /paste:直接貼上序列。 | ||||||
|
3.
|
擷取PC中的序列檔案:點選 "List of files", 再將您電腦中適當的序列檔案輸入即可。 | ||||||
|
|
以下列二條序列為例,選定由 file/database 輸入 輸入序列(1) embl:HSPDGA4 輸入序列(2) embl:HSA238420 |
||||||
|
|
在 required section 設 gap opening penalty =10, gap extension penalty=0.5 | ||||||
|
|
在 Advanced section 選擇scoring matrix (Blossum 62) | ||||||
|
|
在 out put section選擇輸出格式-- (default 為srspair) | ||||||
|
|
注意:needle程式一定要輸入兩條序列;並請記得在執行區右下角選擇batch (背景執行) | ||||||
|
|
按 Go 。 | ||||||
|
|
檢視結果: 點選file, 選擇saved results, 選取本次分析結果,按display。 檢視結果後存檔:在結果畫面左上方點選file, 選擇save to local file, 輸入正確檔案名稱, 按「儲存」,例:存成hspdga4.needle。 |
||||||
|
|
比對結果: 由於 "embl:HSPDGA4" 是 PDGF A chain exon4 的序列,而 "embl:HSA238420" 是 PDGF A chain gene exon1~4 的序列,在結果中很清楚的看到exon 4 對應在A chain 的第7758 至8005位置上。 |
||||||
 |
|||||||
| Water程式: 以local Alignment進行序列的兩兩比對 | |||||||
| water 操作步驟: | |||||||
|
|
Go to :water | ||||||
|
|
輸入序列,有3種方法可供選擇: | ||||||
|
1.
|
由 file /database:鍵入database:accession number。 | ||||||
|
2.
|
copy /paste:直接貼上序列。 | ||||||
|
3.
|
擷取PC中的序列檔案:點選 "List of files", 再將您電腦中適當的序列檔案輸入即可。 | ||||||
|
|
以下列二條序列為例,選定由 file/database 輸入 輸入序列(1) embl:HSPDGA4 輸入序列(2) embl:HSA238420 |
||||||
|
|
在 required section 設 gap opening penalty =10, gap extension penalty=0.5 | ||||||
|
|
在 Advanced section 選擇scoring matrix (default=Blossum 62) | ||||||
|
|
在 out put section選擇輸出格式-- (default 為srspair) | ||||||
|
|
注意:請輸入兩條序列,並記得在執行區右下角選擇batch (背景執行) | ||||||
|
|
按 Go | ||||||
|
|
檢視結果: 點選file, 選擇saved results, 選取本次分析結果,按display。 檢視結果後存檔:在結果畫面左上方點選file, 選擇save to local file, 輸入正確檔案名稱, 按「儲存」。 |
||||||
|
|
比對結果: 由於 "embl:HSPDGA4" 是 PDGF A chain exon4 的序列,而 "embl:HSA238420" 是 PDGF A chain gene exon1~4 的序列,在結果中很清楚的看到exon 4 對應在A chain 的第7758 至8005位置上,由於是local alignment,因此不match的序列就不列出來了,這也正是local alignment和global alignment最大的不同之處。 |
||||||
|
|
|||||||
|
|
|||||||
| 二、多序列並列分析: emma, Jalview | |||||||
| 多序列並列分析通常是用在直接比對兩條或多條序列彼此間相似程度,並且可以將程式計算出最好的排列方式列出。這些程式可以協助使用者比對兩條序列整體的相似度,或是尋找序列間相似區域(Conserved Region),或得知序列間變化較大的區域。比對的結果可以用來判斷一群蛋白質序列間共通的motif,或用來做為分子演化分析的基礎。 | |||||||
| Emma | |||||||
| EMBOSS以emma程式進行多序列並列分析,emma即為ClustalW程式的Java介面,ClustalW程式是常用的多序列並列分析程式,在一群序列間以global alignment的比對方法先進行序列兩兩比對,再將比對好的各序列對以neighbor joining 的方法排定群組(cluster),進行多序列並列分析。由於emma是使用global alignment的比對方法,因此輸入的序列之間必須有一定程度的相似性,否則會無法得到好的結果或是完全無法執行。 | |||||||
| Emma操作方法: | |||||||
|
|
Go to :emma | ||||||
|
|
輸入序列,有3種方法可供選擇: |
||||||
|
1.
|
由 file /database:鍵入database:accession number。或輸入list file“@ + 路徑 + List file”例:@D:\document and settings\users\jade\tf3a.list。List file 的製作方法見隨後本段說明. | ||||||
|
2.
|
copy /paste:直接貼上序列。 | ||||||
|
3.
|
擷取PC中的序列檔案:點選 "List of files", 再將您電腦中適當的序列檔案一一輸入即可。 (i).自行鍵入:打開“記事本”鍵入以下每一條序列的資料庫名稱及accession number,存成純文字檔,記得要存放在正確的目錄中。
SWISSPROT:TF3A_BUFAM (ii). 至資料庫檢索建檔:至SRS 7網站(http://srs.hgmp.mrc.ac.uk/),選擇start a temporary project,進入query頁,先勾選Swissprot資料庫,再在左邊Query forms點選standard, 進入Standard query網頁後,在輸入欄位第一欄下拉式選單選擇"description",然後輸入TFIIIA(需全部大寫),記得消去append wildcards to words,然後按submit query。此時出現結果畫面,共有八比資料,請勾選SWISSPROT:TF3A_BUFAM, SWISSPROT:TF3A_RANPI, SWISSPROT:TF3A_XENBO, SWISSPROT:TF3A_HUMAN, SWISSPROT:TF3A_XENLA, SWISSPROT:TF3A_YEAST等六條序列,在"Perform operation"處勾選 selected only, 然後在View的下拉式選單選擇Names only, 按Save. 在Save網頁選擇Output to "to file(text)",”Save table as ASCII text/table with”,然後在自己的電腦選擇正確的路徑,輸入檔名,(例:tf3a.list)按Save。 |
||||||
|
|
在 required section 設 gap opening penalty =10, gap extension penalty=0.5 | ||||||
|
|
在 Advanced section 檢視並修訂參數。 | ||||||
|
|
Emma所使用的ClustalW 程式對於gap扣分有一些微調的計分選項,例如Gap separation penalty的選項default=8,表示新的gap若位於鄰近的gap八個redidues以內,扣分會較多,以避免短距離內加入太多gap。此外,在Residue specific penalty的選項,則根據已知的蛋白質結構推測,如果一個gap位於 a run of hydrophilic residues, 則扣分較低,因為這個位置可能存在一個loop structure;反之加入gap的位置若沒有 hydrophilic residues,則會根據這個位置一般對結構的重要性來斟酌扣分(residue specific gap propensities)。 | ||||||
|
|
在 out put section輸入Output file name 及Output dendrogram name(可以不用輸入) | ||||||
|
|
注意:請在執行區右下角選擇batch (背景執行) | ||||||
|
|
按 Go | ||||||
|
|
檢視結果: 點選file, 選擇saved results, 選取本次分析結果,按display。 檢視結果後存檔:在結果畫面左上方點選file, 選擇save to local file, 輸入正確檔案名稱, 按「儲存」。 |
||||||
|
|
比對結果: emma 只列出各單ㄧ序列比對後加了gap的結果,因此須先存檔後再用Jalview, showalign, 或prettyplot檢視. |
||||||
 |
|||||||
| Jalview : 檢視編輯multiple sequence alignment的工具 | |||||||
| 由於目前emma只能逐一列出單一序列,無法呈現多條序列並列比對的結果,因此還須使用Jalview 工具來觀察完整的比對結果。。emma比對結果也可輸出dendrogram,用來表示程式在執行並列分析時各序列群組的相似度關係,而非演化樹圖,請注意避免混淆。除了Jalview,JEMBOSS程式組中prettyplot及showalign兩個程式也可用來檢視emma的分析結果,且這兩個程式還可用來列出consensus sequence,會在在一節介紹。 | |||||||
| Jalview 操作步驟 | |||||||
|
|
點選JEMBOSS工作區上方Tools,然後在選單中選擇Multiple sequence editor--Jalview。 | ||||||
|
|
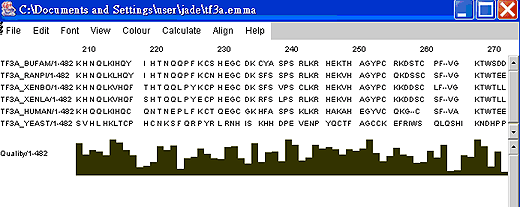
輸入路徑及名稱,例:C:\Documents and settings\users\jade\tf3a.emma 。 | ||||||
|
|
選擇fasta格式 | ||||||
|
|
按launch,畫面會出現多並列分析比對結果. Jalview除檢視比對結果,還可編輯比對結果,並調整色彩。 | ||||||
|
|
存檔。 | ||||||
|
|
比對結果: Tf3a.emma用Jalview呈現的比對結果(部份) |
||||||
|
|
|||||||
|
|
|||||||
| 三、尋找相同序列: showalign, prettyplot | |||||||
| Showalign 及prettuplot這兩個程式可以將多序列並列分析的結果列出,並顯示相同序列(consensus sequence)。其中showalign對於consensus sequence的決定及呈現方式有較多參數可供選擇,prettyplot 則是以default值直接列出consensus sequence。 | |||||||
| showalign: 多序列並列分析結果檢視及列出相同序列 | |||||||
| showalign操作方法 | |||||||
|
|
Go to : showalign | ||||||
|
|
輸入序列,由 file /database輸入msf 檔案,亦即multiple sequence alignment的結果(例:tf3a.emma)。 | ||||||
|
|
點選load sequence attributes | ||||||
|
|
選擇advanced options,設定display格式,如: |
||||||
|
|
列出全部序列:show all sequences | ||||||
|
|
只列出相同序列,不同序列以'‧'代替:Identities between the sequence | ||||||
|
|
只列出不同序列,相同序列以'‧'代替:Non-identities between the sequences | ||||||
|
|
列出相似序列,其他序列以'‧'代替:Similarities between the sequences | ||||||
|
|
只列出不相似序列,其他序列以'‧'代替:Dissimilarities between the sequences | ||||||
|
|
設consensus sequence成立所需的多數值:pluratrity check for consensus | ||||||
|
|
參考序列(the reference sequence )會列在最下方(亦可選擇不列出),可以是consensus sequence (default),或由使用者指定的一條序列。 |
||||||
|
|
比對結果(the output sequences)可依input的順序列出,或照序列相似度、或序列名稱字母順序排列。 | ||||||
|
|
按 GO | ||||||
|
|
檢視結果並存檔。(結果為文字檔,例:tf3a.align) | ||||||
|
|
比對結果: 設定參數show all sequences, 不顯示reference sequences,identities in consensus為大寫字母,show sequence number, show ruler等。Consensus sequence列在序列最上方。 |
||||||
 |
|||||||
| 果設定參數為show non-identities, 顯示reference sequences,則只列出non-identical的序列,並會在下方列出consensus sequence作為reference,結果如下: | |||||||
 |
|||||||
| prettyplot:多序列並列分析的彩色圖示及列出相同序列 | |||||||
| prettyplot操作方法: | |||||||
|
|
Go to : prettyplot | ||||||
|
|
輸入序列,由 file /database輸入msf 檔案,亦即multiple sequence alignment的結果(例:tf3a.emma)。 | ||||||
|
|
點選load sequence attributes | ||||||
|
|
選擇advanced options,設定display格式,記得勾選display the consensus,其餘可接受預設值。 | ||||||
|
|
按 GO | ||||||
|
|
檢視結果並存檔,結果為圖形檔,記得副檔名為 ".png"。 | ||||||
|
|
比對結果: tf3a_pretty1.png(部份)。Prettyplot程式設定只要達total sequence weighting的1/2即可為 consensus sequence。Prettyplot的好處,是可以將多序列並列分析的結果以彩色呈現,並且列出consensus sequence;其最大的缺點是當序列長度較長時,比對結果將被切割成數個檔案來檢視。以tf3aprettyplot.png為例,就需要拆成三個檔案才能將比對結果完全呈現出來。 |
||||||
|
|
|||||||