
最新消息
99 年 2月8-9 日將於國立成功大學醫學院第三講堂舉行「RNA分子生物科技研討會」,歡迎報名參加。
研討會網址:http://www.binfo.ncku.edu.tw/RNA2010/
GMBD Bioinformatics Core 分析工具深度報導
親緣分析的遊戲場:圖像化親緣演化分析工具(POWER)與最佳化分子親緣關係高速分析平台(PALM)
生物巨分子的親緣性分析是一個經常使用的演化分類方法,此方法的主要優點是藉由計算保守性序列的變異程度,而推衍序列之間的相對演變歷史,進一步可推衍為個體間或物種間的族系發生史 (phylogenesis) 。序列親緣性分析可補強型態測量分類方法的不足,除了應用在一般的物種演化分析與生物地理學的應用外外,也可以協助鑑別某些未必具有明顯特徵可資分類的病原菌或病毒株,或是透過序列的變異程度來探知瞭解病原體的傳染途徑與方式。傳統之血清型分類方法,無法有效鑑別病原體變異株之間的些微差異,病源鑑定與疾病控制上亦受限於此;透過代表性基因型或分子標記的定序與後續的序列親緣性分析,我們可瞭解不同時間與地點的採撿樣品之間的時空演變關係,甚至經由同一個體中不同組織器官的病原體序列演化資訊,我們可以得知病原在宿主內的感染路徑,進以掌握致病原變異與傳佈的機制與防制之道。
序列分析在生物資訊的研究領域中已為相當成熟的技術,而分子演化 (molecular evolution) 學者們也成功地引入這些計算理論至生物巨分子序列分析上,發展出利用序列分析估算物種演化的理論。一個標準的親緣性分析包括一連串的操作階段 (如圖一):
1. 多序列排比 (multiple sequence alignment, MSA)
2. 親緣關係推衍 (inferring on phylogenetic relationship)
3. 樹形繪製 (tree building)
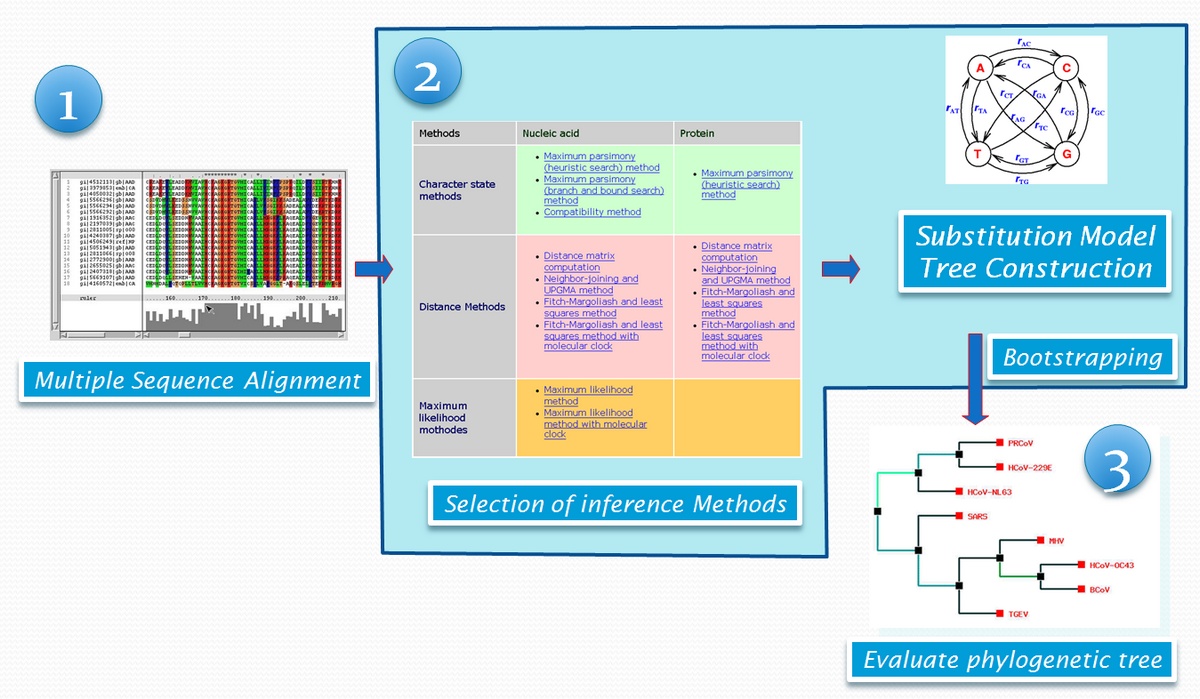
圖一:親緣分析的基本流程
每個步驟都可由個別的程式完成,例如多序列排比 MSA,可透過 ClustalW on command-mode, GUI ClustalX,或其他網路上的生物資訊服務站如 Centre for Molecular and Biomolecular Informatics (http://www.cmbi.kun.nl/)、European Bioinformatics Institute (http://www.ebi.ac.uk)等;親緣關係則以預先完成的序列排比結果為基礎,再透過 PHYLIP package 及 WebPHYLIP 等網路生物資源服務站,或是 PhyML, MrBayes,MEGA 等單機版軟體來進行後續分析。大部分的分析工具都必須挑選合適的演化親緣模型 (Substitution Model),引用不同的演化模型,往往會影響到最後的樹狀結構與生物意義推論 (此一問題將在後面介紹的 PALM 這套系統中獲得解決)。最後的樹形繪製工作,有時已包含在親緣關係分析程式的最後輸出結果之內,如 MEGA;也有些軟體專為樹形繪製設計,可讀取親緣關係分析的輸出檔案 (文字形式的檔案),如 DRAWTREE、WebPHYLIP、 TREEVIEW、NJPLOT、PhyloDraw 與 PHYLIP 套件中的 DRAWGRAM 等。在不同分析階段之間,有許多檔案輸出與輸入格式對應與轉換的問題,而即使有整合性親緣分析服務,如 PhyloBLAST,但是其分析工作的彈性較小,往往無法因應使用者的需求。PhyloBLAST 起始於 BLAST search,僅搜尋蛋白質序列進行親緣分析,但不允許使用者自行調整參數,並以固定的參數與演化模型來建置親緣關係。序列親緣樹形圖是一般生物學及醫學研究者的判讀依據,但在樹形繪製的適切表達上也經常發生問題,例如:樹狀分支間發生交叉,序列識別標記重疊,檔案輸出格式支援性…等等。這些現有的分析工具,往往親和性不足,圖像化使用者介面闕如或不佳,操作過程繁瑣,對於大量或是反覆更改相關參數的親緣分析十分不利,同時也沒有在分析過程中,告知使用者有哪些分析方法與計分矩陣,及何種參數的選擇建議,造成使用者往往徒勞無功,甚至做出錯誤的推論結果。
接下來介紹的這兩個分析平台,將可以讓使用者,跳脫這一切繁雜的操作流程與遠離不同程式間的轉換工作,以圖像化的高親和性介面,透過平滑的學習曲線,並在系統嚴謹的演化統計理論與高效能資訊技術的協助下,快速且正確地建構研究結果所將要呈現的親緣關係,並藉此從中推論出背後所代表的生物意義。
POWER (PhylOgenetic Web Repeater):圖像化親緣演化分析工具
本研究的主要目的,在於建構整合之自動化序列親緣性分析系統,以導引式互動介面協助來自不同研究社群的使用者完成分析,簡化繁瑣的序列格式處理過程及流暢地銜接多種操作程序,但又能保有各分析階段的精細調整參數的功能,並自動記錄操作過程的參數設定,以便使用者回溯其工作流程,再行調整。另外,針對樹形圖的輸出方式,與樹形分支排列調整等功能設計程式,以符合客製化 (customize) 的理想。另外,目前更設計將 POWER 與 BLAST 工具結合,以 GenBank 或是 NR 為比對資料庫,以簡化並延伸使用者的輸入選定序列的工作,加速使用者解析所研究之序列與相關序列間的時空分析概況。
本系統為整合性可反覆執行之序列親緣關係分析工具,提供使用者分別以一般序列 (Fasta) 與比對序列 (Aligned sequences) 二種方法來進行核酸或蛋白質序列親緣關係分析,並可即時動態增加或減少序列數目節點,反覆調整分析之結果 (請參見圖二、三) 。POWER 總共提供 64 種不同組合的分析方法,在每一種分析中,皆以簡單明瞭的指引協助使用者設定選擇參數,在 Help 網頁中更詳述了每個參數的意義,方便使用者即時查看;除了最大概似法 (Maximum Likelihood, ML) 相當耗時之外,大部分分析方法,如距離法 (Distance Matrix Method) 與最大檢約法 Maximum Parsimony) ,都可以數分鐘內完成,使用者可在線上稍做等候,或是將送件完成的網頁存成書籤 (Bookmark) 再次拜訪,同時也可以靜待系統的 E-mail 通知取件。
分析結果中,系統為使用者保留與記錄了所有分析過程中所使用的參數設定,使用者輸入的序列以及運算過程中所輸出的結果,所有檔案皆可在下載區中下載保存;親緣關係樹的呈現上以不同顏色清楚標示可信度的範圍。若要進行另一次的分析,使用者也可以移動滑鼠點擊想要移除的序列所代表的紅色結點,再次重覆以相同或不同的方法進行分析,或新增任何序列,免去不斷重複輸入序列資料與參數的繁瑣過程。點擊黑色結點則可將結點二側的序列反轉,便利使用者呈現其研究結果的呈現序列順序,這樣的調整並不會影響整個親緣樹的原有拓樸架構 (Topology) 。
POWER 系統之建置,乃由國衛院生物統計與生物資訊研究組熊昭組主任、林仲彥助研究員、林凡凱先生、林介華小姐、賴立偉先生及徐秀君小姐,所組成的研發小組以一年半的時間進行開發,並在中央研究院基因體中心幹細胞實驗室陳淑華博士的協同合作下,完成整體系統的優化與論文的撰寫。POWER 已發表於 2005 年七月的國際知名期刊 Nucleic Acid Research (33: W553-W556, 2005) ,同時 POWER 也被多次引用於 Nature Preceding、The journal of experimental biology、Nucleic Acids Research、Bioinformatics、 BMC Bioinformatics 以及 PLoS ONE 等著名期刊的論文之中;並被收錄於知名的蛋白質體學分析工具網站 ExPASy 裡。截自 2009 年 12 月底為止,已有超過 12000 餘人次使用 POWER ,並已處理二十四萬餘條序列。
Citation: Nucleic Acids Research, 2005, Vol. 33, Web Server issue W553–W556
網址:http://power.nhri.org.tw
實例操作:POWER 的操作方法 (step by step)、結果呈現與說明,歡迎觀賞影音檔介紹:
中文版 (Chinese version)
英文版 (English version)
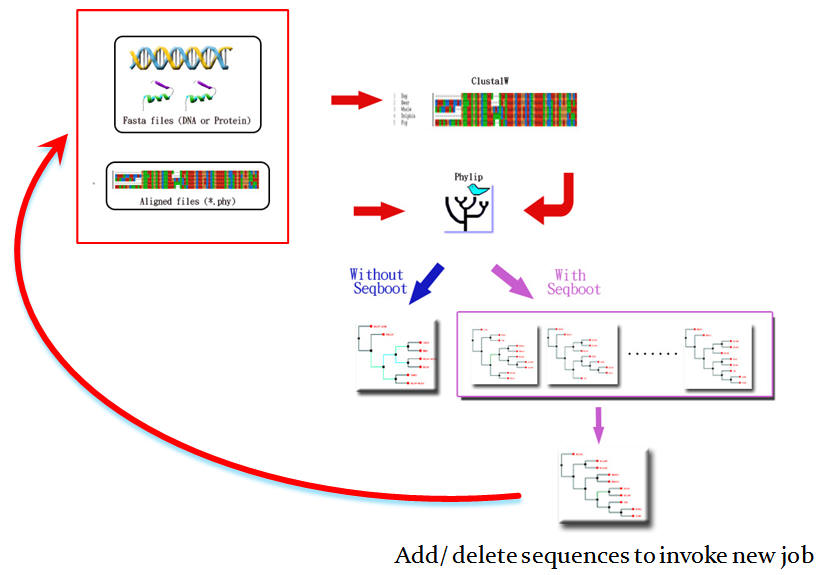
圖二:POWER 分析流程與結果呈現
圖三:POWER 之操作介面
PALM (Phylogenetic Reconstruction by Automatic Likelihood Model Selector):最佳化分子親緣關係分析平台
一直以來,演化模型的選取,都是建構親緣樹的重要關鍵,不同的模型往往會造成樹型拓樸的差異,進而影響後續的生物意義推論。PALM 為一個自動選擇最佳演化模型之生物分子親緣關係分析平台,以最大概似法 (Maximum Likelihood, ML) 為基礎,應用多核心平行技術 (parallel computing),可加速運算效能,縮減所需時間為原來的六分之一 (系統架構如圖四所示)。目前 PALM 提供多達 56 種 DNA 演化模型及 112 種蛋白質演化模型,可同時評估,並依其統計結果擇取最佳模型,再輔以 Bootstrap 分析,進一步提供互動式親緣樹檢視介面,方便使用者對親緣樹型進行翻轉、交換,同時也可以透過圖像化方式刪除與新增分析序列,再次進行新的分析工作。分析流程中所用的參數與產出檔案,都與圖像化結果一起被紀錄在報表網頁中,可供使用者下載並進一步利用 (圖五)。 PALM 以極具親和性的介面,大大地減少使用者在進行複雜多演化模型分析的難度與時間,使得生物學家能以更具統計意義的演化模型,來解釋所建構出來的親緣關係與其中所包含的生物意涵。
PALM 系統之建置,乃由中央研究院資訊科學研究所林仲彥助研究員 (亦為國家衛生研究院群體科學研究所,生物統計與生物資訊研究組合聘之助研究員),所引領的研究團隊 (系統生物與網路生物學實驗室,http://eln.iis.sinica.edu.tw),包括陳淑華博士、蘇聖堯先生、羅存仁先生、郭柏漢先生、陳貴弦先生及黃騰杰先生,所組成的研發小組以兩年半的時間進行開發,並完成整體系統的優化、相關文件與論文的撰寫。PALM 已於 2009 年十二月初發表在國際知名期刊 PLoS ONE (4(12): e8116) 上,截自 2010 年 1 月底為止,已有來自全球超過 900 餘人次使用 PALM,並已處理一萬九千餘條序列。
Citation: PLoS ONE 4(12): e8116. doi:10.1371/journal.pone.0008116
網址: http://palm.iis.sinica.edu.tw
實例操作:Palm 的操作方法 (step by step)、結果呈現與說明
中文版 (Chinese version)
英文版 (English version)
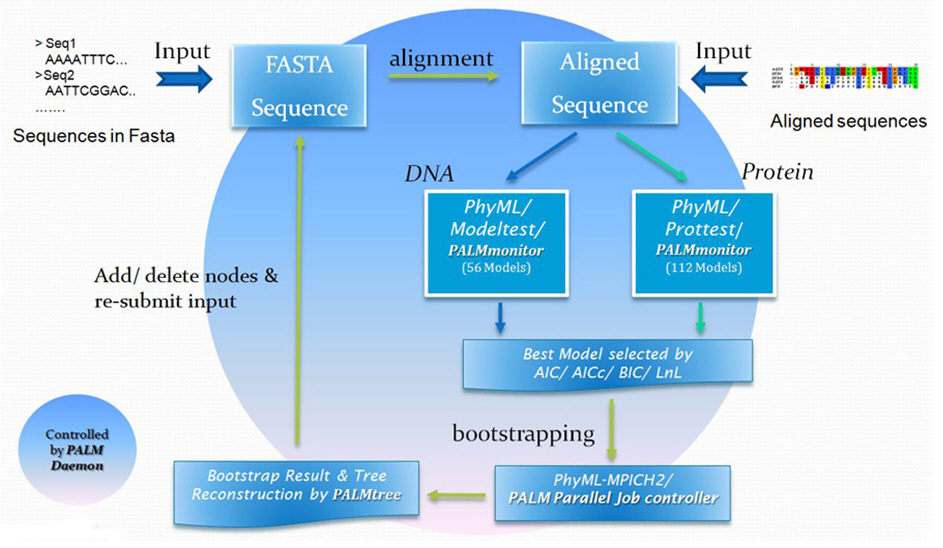
圖四:PALM 的分析流程與基本運作

圖五:PALM 的分析結果與相關檔案之呈現及下載
POWER / PALM 學術研究上的應用範例
POWER 及 PALM 是相當具有親和力的序列親緣分析系統,除了運用於生物巨分子序列分析外,還可以讓研究人員進行疾病病原分子監控,快速判定源自不同時空的檢體序列間的親緣關係,而無須陷入繁瑣的程式操作與避免複雜的結果研判。譬如人類與禽類流感病毒的分析,更可以透過這樣的系統快速地進行親緣與傳遞途徑的解析。
由國家衛生研究院群體科學研究所熊昭所長所帶領的研究團隊,亦利用 POWER 分析來自 1971-2002 年之間 A/H3N2 型流感病毒檢體序列中 hemagglutinin (HA) 的蛋白突變,進一步預測出 1999-2004 年間的病毒株抗原位點 (成功率=91.67%),找出 20 個可能的主要免疫位點。 (Bioinformatics 2008, 24(4):505-512)。

圖六:利用 POWER 分析來自 1971-2002 年之間 A/H3N2 型流感病毒檢體序列中 hemagglutinin (HA) 的蛋白突變。
近期使用者以 POWER 為基礎平台,所發表的學術論文如下:

圖七:以距離矩陣法 (Neighbour-joining mathod) 來分析與非洲肺魚尿素轉運子的相關親緣關係

圖八:以最大檢約法 (Maximum parsimony method) 來分析與魚類促性腺激素受器的相關親緣關係

圖九:以最大概似法 (Maximum likelihood method) 來分析松科植物酵素合成酶親緣關係。
