
最新消息
98 年9月7-8日將於高雄醫學大學舉辦「研究資源2009系列活動:結構生物資訊學研習會」,歡迎報名參加。
報名網址為:http://www.nhri.org.tw/rr2009/
最新發表
整合性高速蛋白質結構搜尋比對伺服器 -- iSARST
iSARST 是一個高速蛋白質結構搜尋比對伺服器,它在多工而平行化的作業環境下整合了數種結構比對工具及兩個高效率的蛋白質結構搜尋方法:一是適合尋找一般結構類似物 (structural homolog) 的 SARST 系統,二是專用於搜尋蛋白質環形序列重組類似物 (circular permutant) 的 CPSARST 系統。
iSARST 允許使用者一次輸入多筆 PDB 或 SCOP 代號 (entry ID),亦可上傳包含許多蛋白質 PDB 結構的壓縮檔案。查詢資料輸入後,伺服器會先以 SARST/CPSARST 快速地搜尋結構資料庫,然後用高準確率的結構比對軟體(如 FAST, TM-align 及 SAMO)重新計算所得蛋白質與查詢蛋白質間的結構相似度並按相似度大小列出結構類似物之清單。透過此兩階段工作模式,iSARST 能快速地回報資料並確保所得資訊的高準確度。其輸出資訊除類似物清單,亦包含各所得蛋白質之功能。此外,iSARST 提供互動式結構檢視介面以利使用者做進一步的結構分析、研究。預期該伺服器可為後基因體時代的結構生物學家們提供方便的工作平台,並可有效運用於新穎蛋白質的功能預測。iSARST 相關論文已於2009年5月刊載於知名國際期刊核酸研究 (Nucleic Acids Research)上,其伺服器網址為:http://sarst.life.nthu.edu.tw/iSARST/
Reference:
1. Lo WC, Lee CY, Lee CC, Lyu PC. (2009). iSARST: an integrated SARST web server for rapid protein structural similarity searches. Nucleic Acids Research, Jul 1;37(Web Server issue):W545-51, Epub 2009.
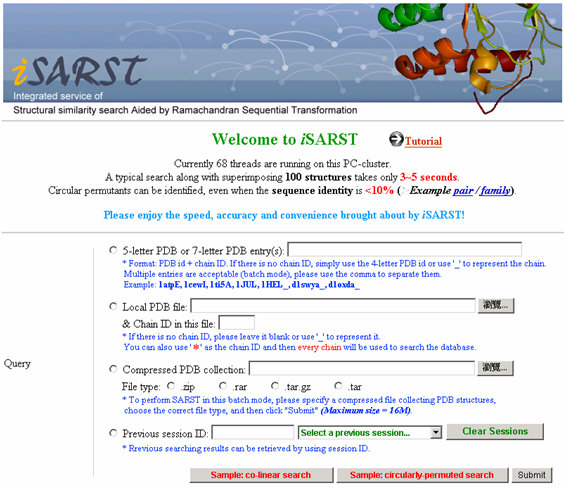 |
|
圖一:iSARST所提供之多種輸入模式 iSARST允許使用者輸入有: |
|
 |
|
圖二:iSARST 操作界面概覽。 (a) 搜尋所得之結構類似物清單。
|
|
分析工具介紹
GEMDOCK -- 虛擬篩選與分析之圖形介面化藥物設計系統
發展一種新藥平均約需花費十二年,成本超過一百億新台幣,這樣耗費大量時間與金錢的傳統新藥開發方式,已無法滿足人類對藥物的需求。針對這個問題,電腦輔助藥物設計提供了一個快速、有效而且低成本的藥物搜尋與分析方法。基於過去的分子嵌合 (molecular docking) 研究和家族競爭演化方法,交通大學楊進木教授實驗室發展出一套以演化式演算法 (evolutionary algorithm) 為基礎的電腦輔助藥物設計軟體:GEMDOCK。
GEMDOCK 是一套整合了自動化分子嵌合、虛擬篩選以及篩選後分析的程式,有使用者十分容易操作的圖形介面,並可即時 (real-time) 將分子立體結構視覺化,提供給使用者觀察。國內有數個實驗室成功應用我們的
GEMDOCK 軟體在不同的藥物標的蛋白上:如 influenza virus neuraminidase (國衛院徐祖安教授)、flaviviruses envelop protein (交通大學楊昀良教授)、幽門桿菌及結核桿菌之shikimate kinase(清華大學王雯靜教授)、CLP-like protein (中興大學周三和教授)bovine beta-lactoglobulin(交通大學毛仁淡教授)、sulfotransferase、imidase(交通大學楊裕雄教授)、geranylgeranyl pyrophosphate synthase(中研院梁博煌教授)、及蛋白質功能設計等。GEMDOCK 下載的網址是:http://gemdock.life.nctu.edu.tw/dock/download.php
Reference:
1. Yang, J.-M. and Chen, C.-C. (2004). GEMDOCK: a generic evolutionary method for molecular docking. Proteins: Structure, Function, and Bioinformatics 55:288-304.
2. Yang, J.-M., Chen, Y.-F., Shen, T.-W., Kristal, B. S. and Hsu, D. F. (2005). Consensus scoring criteria for improving enrichment in virtual screening. Journal of Chemical Information and Modeling 45:1134-1146.
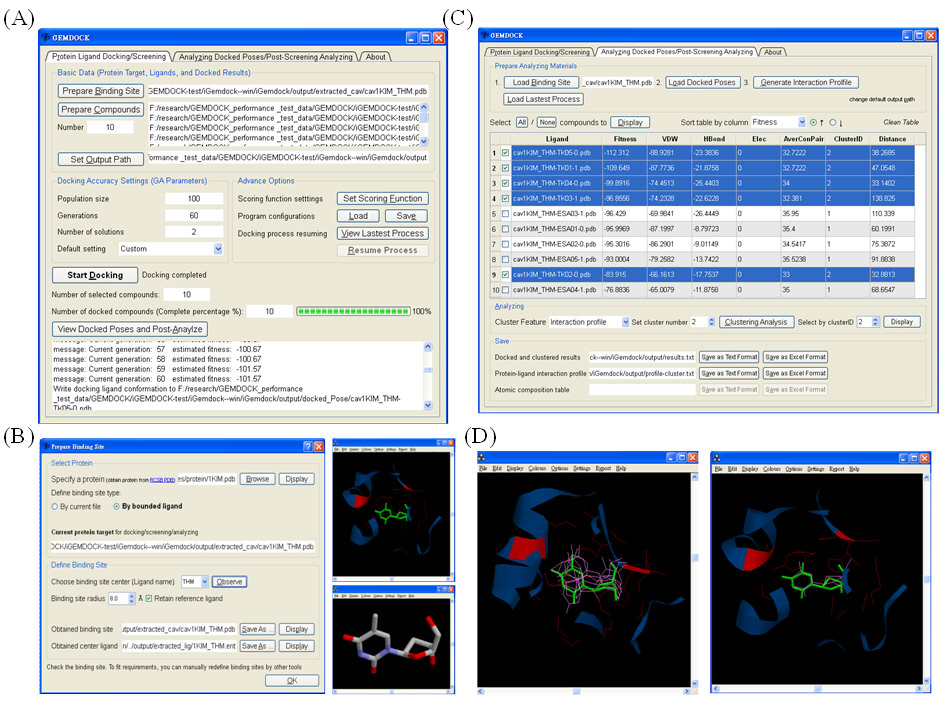 |
|
圖一:iGEMDOCK使用介面一覽(A) 蛋白質與配體嵌合資料之輸入介面,提供使用者輸入各項參數。
|
|
PPISearch -- 蛋白質與蛋白質間交互作用的比對搜尋工具
細胞內複雜的生化網路包含大量的蛋白質-蛋白質交互作用 (protein-protein interactions, PPIs),隨著近幾年實驗技術的進步,國內外線上資料庫已經累積大量的交互作用資料。PPISearch,可以讓使用者輸入一對蛋白質序列或蛋白質名稱,在 PPI 資料庫中快速搜尋與其相似的蛋白質-蛋白質交互作用。此工具所使用的 PPI 資料庫,是我們整合了 IntAct、DIP、MIPS、MINT 以及 BioGRID 五個大型公開資料庫而得來的,包含共計超過29萬筆 PPI 資料。 我們的靈感,來自 BLAST 搜尋相似序列的工具,因此 PPISearch 可視為迅速搜尋相似蛋白質交互作用的 BLAST,能對快速增加但尚不知其功能的 PPIs 提出再進一步研究的線索,這些線索包括生物功能、生化路徑位置、功能性區塊等。這是世界上第一個提出此服務的工具,並已發表於 2009 年的 Nucleic Acids Research 期刊。網址為 http://gemdock.life.nctu.edu.tw/ppisearch/index.php。
Reference:
1. Chen, C.-C., Lin, C.-Y., Lo, Y.-S. and Yang, J.-M. (2009). PPISearch: a web server for searching homologous protein-protein interactions across multiple species. Nucleic Acids Research 37:W376-W383.
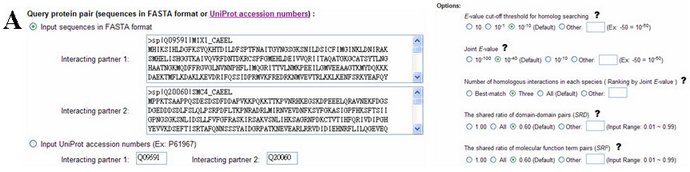 |
|
(A) PPISearch提供使用者輸入兩條蛋白質序列 |
|
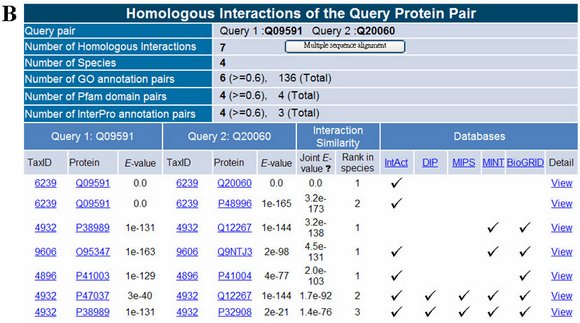 |
|
(B) PPISearch 輸出使用者提問之蛋白質配對的同源蛋白質-蛋白質交互作用,並指出每一筆交互作用是記錄在哪一個公開資料庫中。 |
|
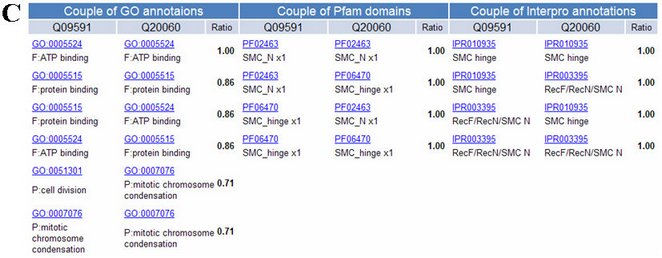 |
|
(C) 輸出使用者提問之蛋白質配對的可能註解 (annotation),提供給使用者做參考。 |
|
3D-partner -- 蛋白質與蛋白質間交互作用的結構結合模式
大部分的生化機制中都涉及蛋白質-蛋白質交互作用。而已有結晶結構的蛋白質交互作用,可以讓研究者了解其交互作用功能區塊 (interacting domain) 以及結合模式 (binding model);對於電腦輔助預測蛋白質交互作用來說,可以提升預測準確度並且提供蛋白質配對的結合模式。3D-partner 從 Protein Data Bank 建立具有 3D 結構的異二聚體 (heterodimer) 蛋白質配對模版資料,再進一步以收集到的模版資料並解釋交互作用的結合模式為何。
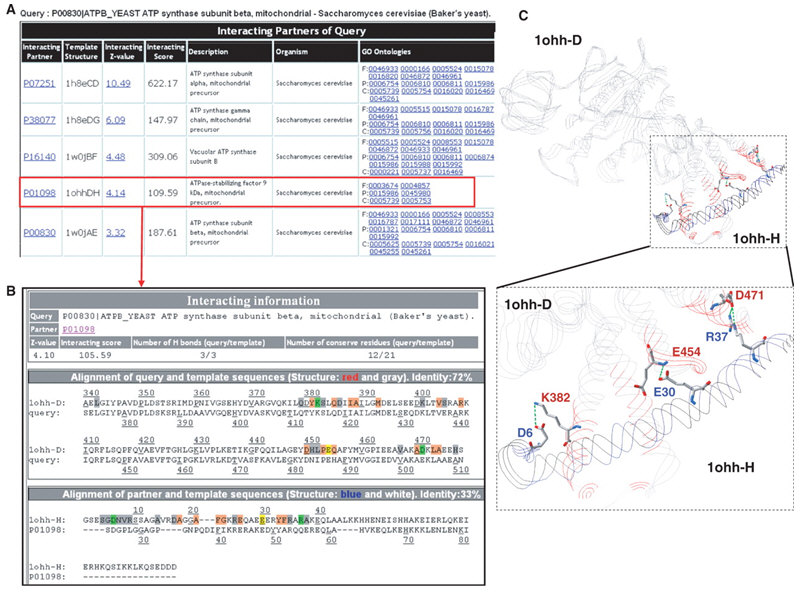
3D-partner 的概念如圖一,先使用 IMPALA 找到輸入蛋白質的同源結構模版 (homologous template),再用 PSI-BLAST 找出該模版的同源蛋白質 (homologous protein),最後透過我們發展的能量函式對蛋白質配對進行算分。根據算分結果,3D-partner 提供使用者預測上統計的顯著性 (Z-score)、結合模式、並指出該模板上重要的胺基酸位置、以及可能的生物功能。3D-partner 已在 2007 年發表於 Nucleic Acids Research,網址是:http://3d-partner.life.nctu.edu.tw。
Reference:
1. Chen, Y.-C., Lo, Y.-S., Hsu, W.-C. and Yang, J.-M. (2007). 3D-partner: a web server to infer interacting partners and binding models. Nucleic Acids Research 35:W561-567.
 |
|
(A) 列表呈現預測會與酵母菌ATP2交互作用的蛋白質。
|
|
蛋白質同源結構之快速搜尋工具 -- 3D-BLAST
進入後基因體時代,功能性結構基因體學的研究愈來愈重要。基因體計畫中大部分的基因所轉譯之蛋白質,透過實驗技術已能快速解出其結晶結構,但尚無法立刻明瞭其生物性功能為何。針對這議題,我們以結構字元(structural alphabet) 集構編碼與 BLAST 快速搜尋為核心,發展出快速搜尋、比對蛋白質結構資料庫的工具:3D-BLAST。此一工具可應用在同源蛋白質搜尋 (homology searching) 及功能性分類 (fold assignment) 等重大生物議題。當一個新的蛋白質結晶結構發表後,研究者尚不知該蛋白質的分類或功能,可經由 3D-BLAST 快速搜尋結構資料庫,決定其功能性演化的分類,並進一步推測它的生物功能。3D-BLAST 搜尋資料庫時,不需要真正疊合蛋白質立體結構,因此可在2秒內快速搜尋一萬五千個以上的蛋白質三級結構。此外,如同 BLAST 在序列比對後會提供具有統計意義的 E-value,3D-BLAST 也能在搜尋結構時提供此統計閾值。結果顯示,當 E-value 小於10-15 時,3D-BLAST 在功能性片段分類的準確度可達到 98%。目前 3D-BLAST 之相關研究已發表在 Nucleic Acids Research 與 Genome Biology 等國際期刊,網址為:http://3d-blast.life.nctu.edu.tw/。
Reference:
1. Tung, C.-H., Huang, J.-W. and Yang, J.-M. (2007). Kappa-alpha plot derived structural alphabet and BLOSUM-like substitution matrix for fast protein structure database search. Genome Biology 8:R31.1-R31.16.
2. Yang, J.-M. and Tung, C.-H. (2006). Protein structure database search and evolutionary classification. Nucleic Acids Research 34:3646-3659.
3. Tung, C.-H. and Yang, J.-M. (2007). fastSCOP: a fast web server for recognizing protein structural domains and SCOP superfamilies. Nucleic Acids Research 35:W438-W443.
 |
|
(A) 網站首頁。使用者可輸入PDB id、SCOP id 或自行上傳蛋白質結構來作搜尋。 |
|
