最新發表
解析複雜生物網路的好工具 -- Hubba
Hubba (Hub Objects Analyzer) 線上網路樞紐分析器,用以解析複雜生物網路交互作用系統中的重要樞紐蛋白質;透過高親和性的使用者界面,研究者可以將欲進行分析的小規模或大量的生物交互作用資料以PSI、Tab格式上傳,並在極短時間內便可呈現出圖形化結果與提供分析資料下載。本系統整合二種由研究團隊所研發的特徵分析演算法則:Maximum Neighborhood Component (MNC) 與 Density of Maximum Neighborhood Component (DMNC),此二種演算法乃是發展用於辨識交互作用網路中的樞紐或關鍵模組,其分析效能比常用的degree 與 bottleneck 分析法來的更為精確,除了 MNC, DMNC 外亦提供 degree, bottleneck 等常用方法的結果;計算所得之重要蛋白質節點,將會以圖像化手法加以呈現,以不同色溫的深淺來呈現其綜合計分數值,並顯示這些重要結點之間的關連性;此外,系統將直接交互作用關係與間接交互作用網路,分別以實線和虛線來表示,藉此可以彰顯這些蛋白質之間的互動關係。研究者更可進一步利用附加選項來瀏覽樞紐蛋白質周邊,以最短路徑相連的其他結點,亦可以輸入特定的蛋白質表列,Hubba 會把這些成員所形成的次網路,由整個複雜網路中擷取出來,並找出與這些蛋白質調控相關的未知成員。
Hubba 網址: http://hub.iis.sinica.edu.tw/Hubba/ 以 Hubba 分析 IntAct 生物分子交互作用資料庫中的交互網路結果:
http://hub.iis.sinica.edu.tw/Hubba/Help/main.htm#Analysis簡介影片:http://hub.iis.sinica.edu.tw/Hubba/Demo/tutorial.htm
Reference:
1. Chung-Yen Lin, Chia-Hao Chin, Hsin-Hung Wu, Shu-Hwa Chen, Chin-Wen Ho, Ming-Tat Ko. Hubba: hub objects analyzer—a framework of interactome hubs identification for network biology. Nucleic Acids Research 2008, doi:10.1093/nar/gkn257 (Web Server Issue).
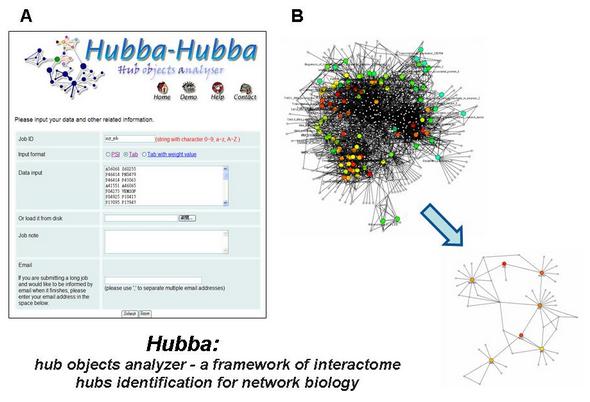 |
圖一: (A) Hubba 分析工具網頁、 (B) Hubba 能辨識複雜生物 |
分析工具介紹
尋找 SNP:NCKU SNP 加值型資料庫
Single nucleotide polymorphism (SNP) 是遺傳上最常見的基因變異;我們主要目地在建立一個加值型的資料庫,其中不只將兩個主要的 SNP 資料庫 (dbSNP及HGVbase) 的資料經過剖析後存入我們的資料庫中,更新增了一個專屬華人族群的 SNP 資料庫,將這三者結合,形成了一個整合性的SNP資料庫。我們已經將網站介面全面改版為一個更具親和力及容易使用的網站介面,方便使用者更容易利用此介面來操作所需要的功能,及利用文字或序列搜尋來得到一個單獨的或整合性的結果 (圖一) ,網站並有提供年、月、總人數的統計資料,及現在所使用的資料庫版本、出版日期及網站更新日期,並提供使用者將已建立好的 SNP 資料轉換成 NCBI 可接受的格式,方便使用者直接將檔案送至 NCBI 建置新的資料。現階段我們已經開放使用者來申請使用 SNP 資料登錄功能,及資料搜尋功能。未來我們希望透過這些持續增加的 SNP 資料,尤其是華人的資料,來更進一步的研究基因與疾病之間的關聯性。NCKU SNP 加值型資料庫的首頁及其網址如圖二 (http://www.binfo.ncku.edu.tw/snp/)。
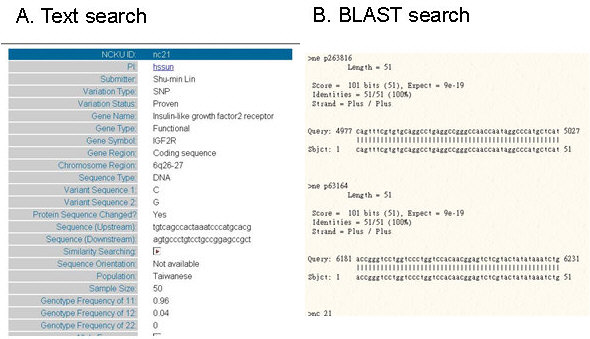 |
圖一:NCKU SNP 加值型資料庫的文字或序列搜尋結果,A 為 TEXT Search、B 為 BLAST Search 結果 |
 |
圖二:NCKU SNP 加值型資料庫首頁,網址為:http://www.binfo.ncku.edu.tw/snp/ |
POWER:生物巨分子序列親緣分析流程之整合與最佳化系統
POWER (PhylOgenetic WEb Repeater) 為一自動化序列親緣性分析系統,以導引式互動介面協助來自不同研究社群的使用者完成分析,簡化繁瑣的序列格式處理過程及流暢地銜接多種操作程序,但又能保有各分析階段的精細調整參數的功能,並自動記錄操作過程的參數設定,以便使用者回溯其工作流程,再行調整。另外,針對樹形圖的輸出方式,與樹形分支排列調整等功能設計程式,以符合客製化(customize)的理想。另外,未來將與 BLAST 工具結合,以 GenBank 或是 NR 為比對資料庫,以簡化並延伸使用者的輸入選定序列的工作,加速使用者解析所研究之序列與相關序列間的時空分析概況。
本系統為整合性可反覆執行之序列親緣關係分析工具,提供使用者分別以一般序列 (FASTA format) 與比對序列 (Aligned sequences) 二種方法來進行核酸或蛋白質序列親緣關係分析,並可即時動態增加或減少序列數目節點,反覆調整分析之結果。
如 POWER 這般具親和性的序列親緣分析系統,除了運用於生物巨分子序列分析外,還可以讓研究人員進行疾病病原分子監控,快速判定源自不同時空的檢體序列間的親緣關係,而無須陷入繁瑣的程式操作與避免複雜的結果研判,譬如人類與禽類流感病毒的分析,更可以透過這樣的系統快速地進行親緣與傳遞途徑的解析。POWER 網址:http://power.nhri.org.tw
| Demo flash for POWER in English/Chinese: http://hub.iis.sinica.edu.tw/Hubba/Demo/tutorial.htm |
Reference:
1. Chung-Yen Lin, Fan-Kai Lin, Chieh Hua Lin, Li-Wei Lai, Hsiu-Jun Hsu, Shu-Hwa Chen, Chao A. Hsiung. POWER: PhylOgenetic WEb Repeater — an integrated and user-optimized framework for biomolecular phylogenetic analysis. Nucleic Acids Research (2005) 33:W553-W556; doi:10.1093/nar/gki494
(Web Server Issue).
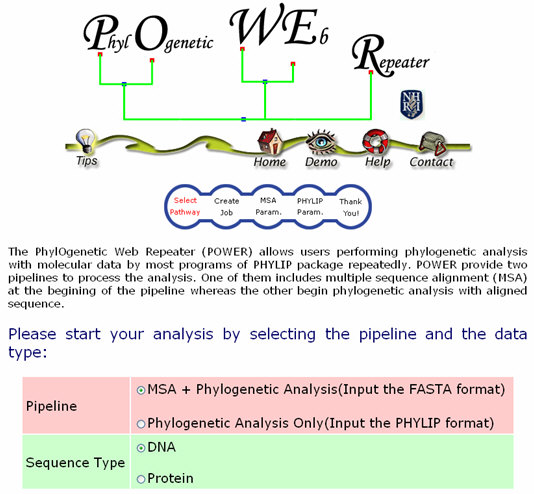 |
圖一: POWER 網站介面。分析方式有 1) 序列比對 + 親緣演化分析,以及 2) 單純的 親緣演化分析兩種方法。使用者可以選擇以 DNA 或 Protein 序列進行分析。 |
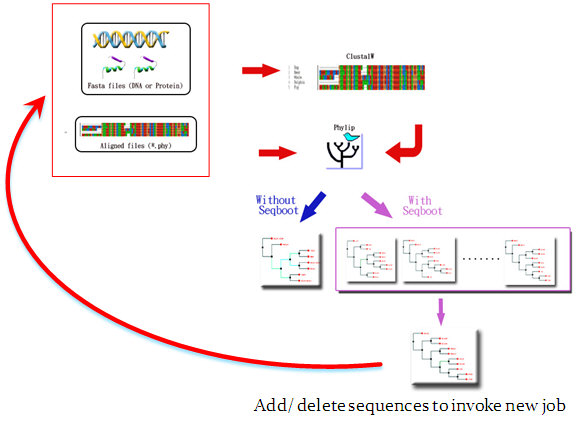 |
圖二:POWER 為整合性且可反覆執行之序列親緣關係分析工具,使用者 可即時動態增加或減少序列數目節點,反覆調整以獲得最佳之分析結果。 |
TAG:尋找癌症關聯基因的加值資料庫
癌症 (Tumor) 為目前十大死因之一,TAG(Tumor Associated Gene)資料庫的建立主要的目的在利用電腦計算搜尋的策略,來辨認新穎的癌症相關基因。第一階段的研究中,我們利用 PubMed 資料庫透過資料探勘的方式來尋找及辨認已知的致癌及抑癌基因。然後再利用一個半自動的資訊擷取系統,到特定資料庫收集目標基因的特定資訊,並儲存到 TAG 資料庫中。第二階段,我們設計便利的網頁介面,提供方便的功能幫助使用者搜尋 TAG 資料庫裡的資訊及分析蛋白質可能與癌化相關的資訊。第三階段,利用所收集到的癌症相關蛋白質之功能性區域資訊,建立加權值量表。最後,藉由搜尋比對人類資料庫中完整的 cDNA 序列,配合已建立的功能性區域之加權值量表,來辨認出新穎的癌症相關基因。TAG 資料庫目前已收集 519 筆癌症相關基因的資料, 其中包括 198 致癌基因, 170 抑癌基因及 151 參與在癌症生成相關基因。 自 2006 年 5 月至今, TAG 資料庫已吸引 26,639 人次使用,使用者更是遍佈全球。
Reference:
1. Chan HH, Tsai SJ, Sun HS. TAG: a comprehensive database for cancer research. (2007) Nucleic Acids Research Molecular Biology Database Collection # 922.
 |
圖一: TAG 資料庫的首頁,網址為:http://www.binfo.ncku.edu.tw/TAG/ |
 |
圖二: TAG 資料庫使用者廣泛分佈於全球各地。自 2008 年 9 月 19 日至今,即有 333 人次使用 TAG 資料庫查詢相關資訊。 |
研討會資訊
第二屆 RNA 分子生物科技研討會 (RNA Symposium 2009)
RNA 相關領域的研究近年來的發展迅速,從早期對 rRNA、tRNA、mRNA 的轉錄及調控機制的研究,到mRNA splicing 的發現與探討,近年來關於 mRNA 的代謝調控、translation 的調控機制之研究,因著 RNA interference、 nocoding RNA、microRNA 的發現,顯示 RNA 在生物體內的角色是越發重要,也開啟對於 RNA 如何調控基因表達的功能有了更不一樣的認識。越來越多的文獻資料顯示這些RNA的調控機制扮演了一個很重要的角色。若其中一種機制的調控發生變化後,許多基因的訊息核醣核酸表現也會因而改變,使的蛋白質表現改變而影響了細胞的生理反應。研究報告也顯示訊息核醣核酸代謝調控的異常會導致疾病的形成如;癌症 (cancer)、慢性發炎 (chronic inflammatory response) 與心血管疾病 (coronary disease),此領域的研究也是後基因體時代的一個越來越重要的研究領域。
在國際社會相關於 RNA 方面的研究每年都會經由 The RNA society 所舉行的年會來促進會員間的彼此交流,近年來與會人數不斷增加,也促進了 RNA 相關領域的研究。有鑑於國內從事RNA相關研究學者日益增加,為了使國內的研究學者有更多交流的機會、以快速提昇國內 RNA 的相關研究、並增加學生對 RNA 相關科學的瞭解與認識,本中心於 2008 年開始籌辦「RNA生物科技討論會」,利用學術討論會的模式,藉著邀請國內外學者的演講與討論來達到學術研究提升的目的。今年舉辦第二次研討會,於2月14日在成功大學舉辦。除邀請國內外學者共 11 名進行演講外,還有 28 篇壁報參展。研討議共吸引近 250 名學員的參與,可說是一個很成功的會議 (相關資訊請參考網頁 http://www.binfo.ncku.edu.tw/RNA2009/) 。
 |
| 圖一:第二屆 RNA 分子生物科技研討會演講情形,參與人數眾多。 |
 |
| 圖二:與會者熱烈討論 POSTER 並相互交換研究心得。 |
分析工具教學
Primer Design Assistant, PDA -- PCR 核酸引子設計線上輔助系統操作教學
快速的診斷為治療疾病的重要工具,也是大規模檢測與研究所亟需。有鑒於現今常用的 PCR 核酸引子設計程式,所套用的模型往往過於簡化,而無法找到最合適的引子配對,也無法因應 High throughput 大規模實驗(譬如 Microarray 相關研究)所需。本系統透過圖像化介面網站,容許一次單筆或多筆資料同時進行線上最佳化分析,提供核酸引子設計諮詢。其演算法設計考量因子包括 GC content, Tm, secondary structure, ion strength, G/C clamp, repeat sequences 及是否包括特定序列區塊等,並整合 thermodynamic theory 及實驗室經驗,以模擬真實狀況下 primer/ template 之間的雜合反應與專一性,為每一條序列找出最佳的引子對。經實驗室驗證,PDA 所設計出的核酸引子可有效提高實驗效能與成功率;本系統更可應用於 General PCR 與 Nested PCR 的核酸引子設計,加速病毒檢體偵測試劑的開發,利用此一系統的協助下,已成功開發多個診斷試劑。譬如於 2003 年中所開發出 SARS-CoV 的快速 nested PCR 檢測方法,便能在一個多小時內對低病毒量樣本進行鑑別,快速判別受感染與否。本系統由 2003 七月起提供服務,截自 2008 年底,共有來自全球各地約十一萬五千人次上線使用,共處理近六十七萬條序列,演算法論文與網站已發表於 Nucleic Acids Research 2003 Web Application Issue,並收錄於多個重要生物資訊工具網站之中。 PDA 的網址為: http://dbb.nhri.org.tw/primer/index.html
| Demo flash for PDA in English/Chinese: http://dbb.nhri.org.tw/primer/help.html |
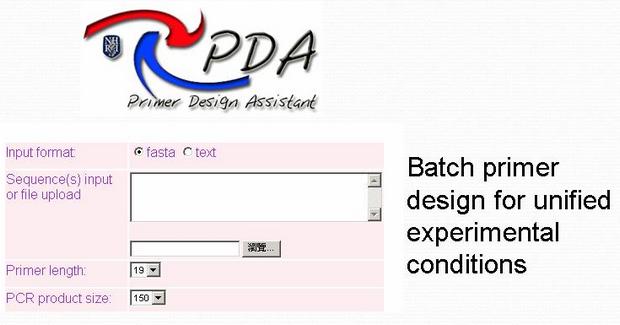
step1: 連至 PDA 網站,輸入核酸序列以搜尋引子 (Primer)
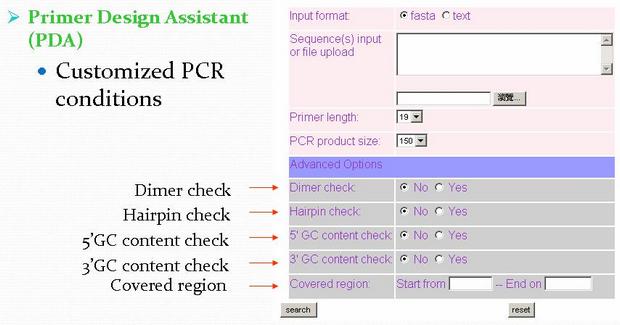
step2: 使用者可以自行調整參數之選項
step3: PDA 分析後的結果,並列出合適的引子對 (Primer)
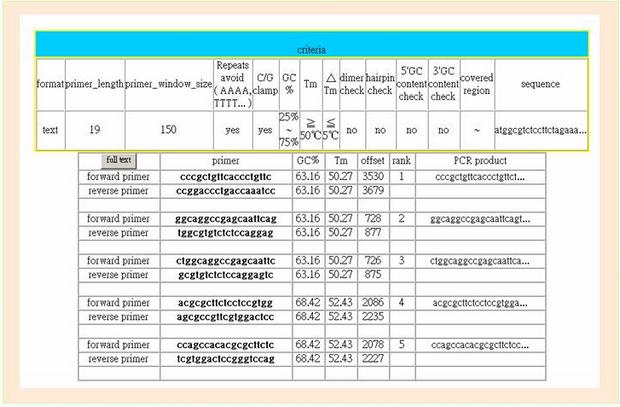
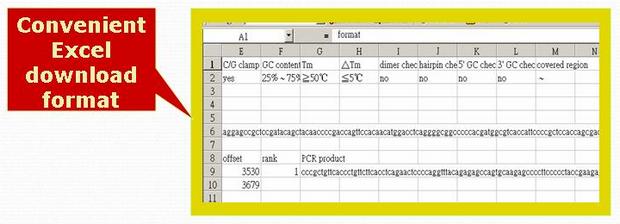
step4: 分析結果可利用 Microsoft EXCEL 格式儲存下來
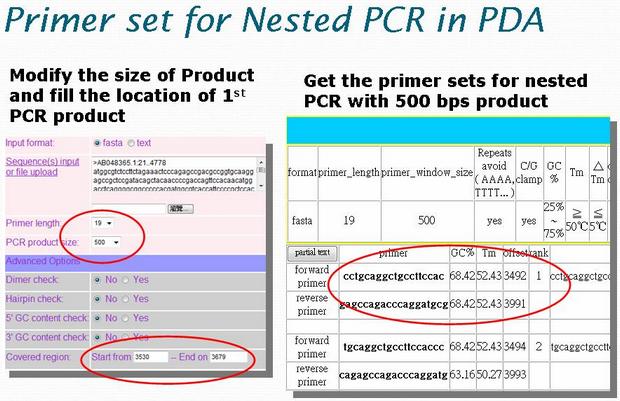
step5: PDA 也能為 Nested PCR 方法設計合適之引子
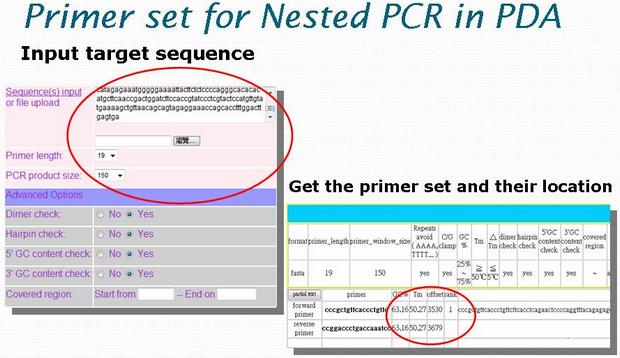
step6: Nested PCR 方法設計引子的結果