
基因體外顯子組定序序列變異之分析流程
Analytic pipelines for the identification of sequence variations in exome sequencing
● 外顯子組序列變異之重要性
外顯子組(exome)為生物基因組中所有蛋白質表現區域(外顯子, exons)之總和。以人類基因組舉例而言,exome包含約十八萬個exons,具有三千萬個鹼基的exome雖然僅占全基因組(三十億個鹼基)的1%,目前由DNA突變引起的疾病估計有85%以上來自於exome區域的序列變異[1,2]。因此次世代基因體定序技術中,外顯子組定序(exome sequencing, 如圖一)遠比進行全基因組序列測序更高效且經濟,其目標區域覆蓋度也更高,利於變異檢測。在臨床研究及應用上,對於未知原因遺傳性疾病或複雜性疾病已開始利用exome sequencing尋找致病基因進而發展治療方法,未來以其大量篩檢與疾病有關基因已成為可能。
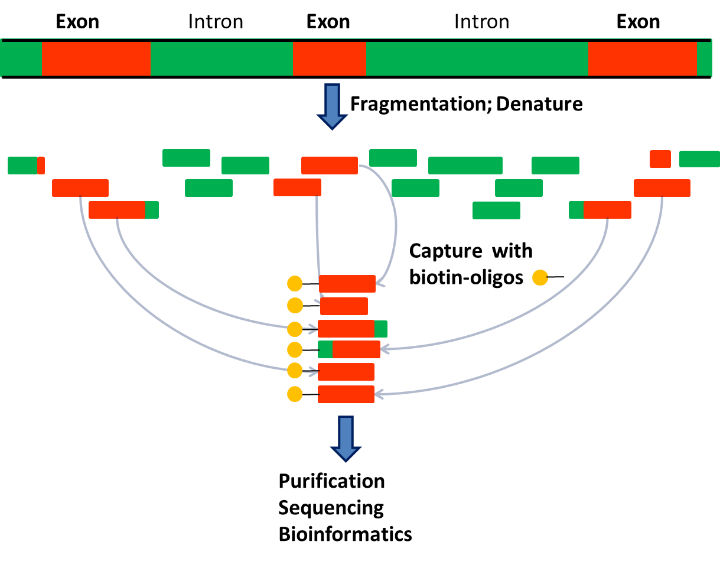 |
圖一. 外顯子組定序流程 (1)雙股DNA片段化及解離為單股DNA (Fragmentation; Denature). (2) 以標記探針捕捉外顯子片段(Capture with biotin-oligos). (3) 純化、定序、及生物資訊資料分析(Purification Sequencing and Bioinformatic analysis). |
|
● Exome sequencing資料分析流程之全面性與完整性
外顯子組序列變異之種類可包括:單核甘酸變異、插入或刪除短片段變異、及拷貝數變異(SNVs, InDels and CNVs)等。利用系統性的分析與彙整、文獻中新發展的生物資訊分析方法、線上最新的資料庫工具之搜尋與比對,E2 TBI(Taiwan Bioinformatics Institute )台灣生物資訊核心設施(簡稱E2 TBI核心)孫孝芳教授團隊,建立了全面且完整的外顯子組序列變異分析流程(如圖二) 。分析步驟包含了基本的序列比對(Map)、發現序列變異 (Variant Analysis),以及進一步深入探討序列變異可能的重要性與功能性、或其生物意義為何(Variant Annotation and Functional Analysis) 。此分析流程已成功地應用於研究實例中,研究成果亦發表於今年的Biomarkers and Genomic Medicine期刊中[3] (如圖三),此分析流程屬於E2 TBI核心客製化服務項目重點之一,未來我們將持續地研發並更新資料庫,期望幫助更多研究者。
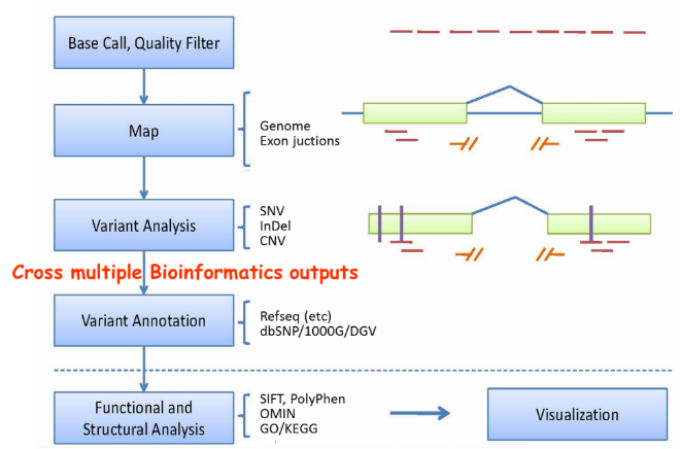 |
圖二. 外顯子組序列變異分析流程 (1)與參考基因組序列比對 (Map). (2) 變異分析(Variant Analysis). (3) 與資料庫比對分析(Variant Annotation). (4) 功能性分析(Functional Analysis). *Refseq: NCBI Reference Sequence Database. dbSNP: SNP database; 1000G: 1000 Genomes; DGV: Database of Genomiv Variants (CNV collection). SIFT, PolyPhen: databases for SNV functional prediction. OMIN: Catalog of Human Genes and Genetic Disorders; GO: Gene Ontology; KEGG: database of metabolic pathways |
|
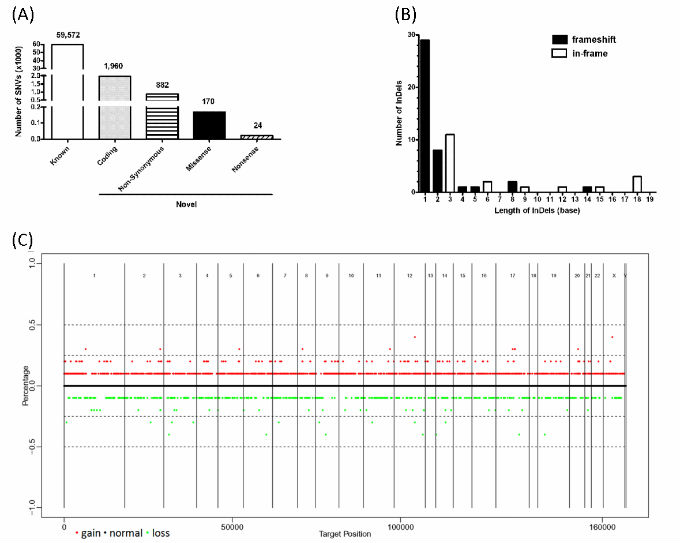 |
圖三. 台灣漢民族外顯子組序列變異偵測分析 * (A)SNV; non-synonymous: 不同義取代; missense: 錯義突變(使密碼所對應的氨基酸改變); nonsense: 無義突變(使原本可製造蛋白質的密碼變成停止密碼); (B)InDel; frameshift: 移碼突變(非三之倍數); in-frame: 符合讀框的(三之倍數); (C)CNV; red: gain; green: loss; black: normal [3] |
參考文獻
1. Ng SB et al. "Targeted capture and massively parallel sequencing of 12 human exomes". Nature 2009. 461 (7261): 272–276. doi:10.1038/nature08250
2. Choi M et al. "Genetic diagnosis by whole exome capture and massively parallel DNA sequencing". PNAS 2009. 106 (45): 19096–19101. doi:10.1073/pnas.0910672106
3. Lin YC et al. “Comprehensive analysis of coding sequence variation in Taiwanese Han population”. Biomarkers and Genomic Medicine 2014. In press (published online 26 June 2014). doi:10.1016/j.bgm.2014.05.001