
創新的演化學習用於蛋白質功能預測
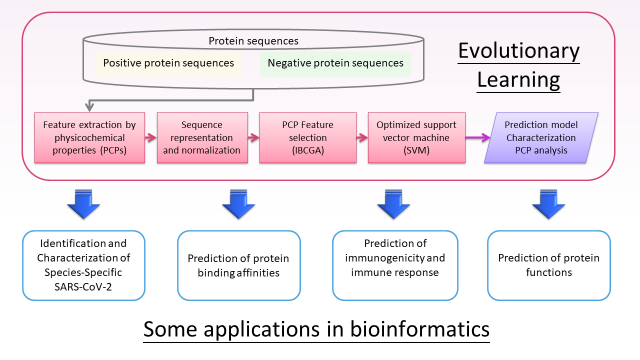
圖1、系統化的演化學習方法應用於蛋白質功能預測。本演化學習基於雙目標組合最佳化演算法使用AAindex數據庫擷取蛋白質序列的PCP特徵,並挑選最少具有鑑別力的特徵進行最佳化建模預測與分析。
近年來國際間新興疾病發生頻仍,面對像新型冠狀病毒(COVID-19)快速傳播的疫情,對於病原體精準且深入的了解需求迫切,以便設計出疫苗和治療方法,其中蛋白質及胜肽的功能預測成為其中重要的研究議題。氨基酸序列是決定蛋白質結構並執行功能的重要基礎,而胺基酸的物理化學特性(physicochemical properties, PCPs)也成為判斷蛋白質特性的寶貴特徵。經過大量的實驗和理論研究,目前計有566個PCPs收錄於AAindex數據庫中。
本核心國立陽明交通大學生物資訊及系統生物研究所何信瑩教授所帶領的研究團隊發展了一套系統化的方法,基於演化學習(Evolutionary learning)及支持向量機(Support vector machine, SVM)的分類器來預測和分析蛋白質功能。演化學習主要採用智慧型演化式演算法對資料導向之機器學習做系統參數最佳化的預測系統,能夠在有限的訓練樣本中辨識一組最小生物標記(signature)及最高預測率的數學模型。要從大量的n個候選生物標記中找出一小組m個生物標記,其中m不為已知,並要達到最佳的預測效能,這是一個C(n, m)的組合最佳化問題(combinatorial optimization problem)。本演化學習技術發展出辨識一組生物標記集合的雙目標組合最佳化演算法IBCGA,同時考慮因為訓練樣本不充分而導致的欠定(underdetermined)問題,希望找出最強健的一組生物標記集合。
本演化學習基於雙目標組合最佳化演算法能自動辨識AAindex數據庫中的一組富含鑑別力的PCPs,同時考慮預測準確性和特徵的機制分析,協助研究蛋白質的生物化學特性及未知蛋白質的功能預測。本核心技術於蛋白質相關的應用廣泛,諸如蛋白質所屬物種的預測、蛋白質結合目標及強度的預測、蛋白質的功能預測、甚至免疫反應中表位(epitopes)的篩選……等。本演化學習方法能夠協助研究人員對龐大的蛋白質進行快速篩選,加速研究及生技開發進程。本文對此演化學習目前提供生醫AI建模的研究服務做一簡介,系統化的演化學習方法除了先前介紹的miRNA生物標記特徵在癌症的預測系統應用外,應用於蛋白質功能預測的方法流程可參考圖1。
1.蛋白質所屬物種預測
由於COVID-19的傳播率不斷提高,迫切需深入闡明其潛在機制,以便可以設計出疫苗和治療方法。先前研究發現SARS-CoV-2與蝙蝠和穿山甲病毒具有遺傳相似性,但對其蛋白質在氨基酸序列及功能缺乏全面的了解。本研究團隊使用演化學習從4320種冠狀病毒的蛋白序列辨識一組人類和非人類冠狀病毒的11個物理化學特性(例如結合自由能),建立一套能夠針對物種特異性冠狀病毒進行分類的辨識系統。在下降蛋白質序列相似性到90%,可獲得97.80% 獨立測試的準確率,並期望透過分析PCPs協助SARS-COV-2藥物結合位點的預測[1]。
2.蛋白質結合親和力預測
蛋白質-蛋白質相互作用(Protein-protein interactions)涉及各種生物學過程,並且相互作用的潛在機制在治療和蛋白質工程中起著至關重要的作用。此研究利用演化學習以及SVM,僅從序列中預測異二聚體蛋白複合物(heterodimeric protein complexes)的結合親和力,並基於結合親和力的預測來對異二聚體蛋白複合物進行分類取得83.33% 獨立測試的準確度,優於其它的機器學習演算法[2]。
3.免疫反應預測
主要組織相容性複合物(MHC)結合在免疫原性(Immunogenicity)[3]以及T細胞的免疫反應(T-cell reactivity)[4]預測都是開發病原體疫苗和癌症疫苗的輔助設計都是很重要的研究議題,也是免疫資訊學的一個目標。本核心技術能夠同時優化特徵選擇和分類器設計,最大程度地提高預測精度,開發第一個出以物理化學性質使用與人類I類MHC分子相關的表位來預測肽免疫原性的計算系統。本研究的成果將能夠進一步應用於人類疫苗、動物疫苗以及癌症疫苗開發中。
4.蛋白質功能及位點預測
蛋白質的折疊、翻譯後修飾(post-translational modification, PTM)及結合位點……等將決定其功能的執行,因此對於這些蛋白質特性的預測是相當重要的議題。本研究團隊利用物理化學作為蛋白質/胜肽的特徵,基於演化學習方法搭配SVM或支持向量回歸(SVR),找到一組有辨別力且穩定的PCP進行相關議題的功能預測。本演化學習方法能夠有效的辨認與DNA結合結構/領域(domains)[5]、辨識特定位點的PTM[6]、預測蛋白摺疊區域[7]……等十餘篇期刊論文。
參考文獻
- Yerukala Sathipati, S., Ho, S.-Y.*, Identification and Characterization of Species-Specific SARS-CoV-2 Physicochemical Properties, Preprint from SSRN, 24 Sep. 2020, DOI: 10.2139/ssrn.3677481 PPR: PPR240467.
- Yerukala Sathipati, S., Wang, J.-R., Hsu, K.-T., Tsai, M.-J., Charoenkwan, P., Huang, W.-L., Ho, S.-Y.*, Characterizing informative sequence descriptors and predicting binding affinities of heterodimeric protein complexes. BMC Bioinformatics, 16(S18), 1-11, 2015.
- Tung, C.-W., Ho, S.-Y.*, POPI: predicting immunogenicity of MHC class I binding peptides by mining informative physicochemical properties, Bioinformatics, 23(8), 942-949, 2007.
- Tung, C.-W., Ziehm, M., Kämper, A., Kohlbacher, O., Ho, S.-Y.*, POPISK: T-cell reactivity prediction using support vector machines and string kernels. BMC Bioinformatics, 12(1), 1-11, 2011.
- Huang, H.-L., Lin, I.-C., Liou, Y.-F., Ho, S.-Y.*., Predicting and analyzing DNA-binding domains using a systematic approach to identifying a set of informative physicochemical and biochemical properties. BMC Bioinformatics, 12, S47, 2011.
- Wang, J.-R., Huang, W.-L., Tsai, M.-J., Hsu, K.-T., Huang, H.-L., Ho, S.-Y.*, ESA-UbiSite: accurate prediction of human ubiquitination sites by identifying a set of effective negatives. Bioinformatics, 33(5), 661-668, 2017.
- Liou, Y.-F., Huang, H.-L., Ho, S.-Y.*, A hydrophobic spine stabilizes a surface-exposed α-helix according to analysis of the solvent-accessible surface area. BMC bioinformatics, 17(19), 171-182, 2016.