
Kart: 一個以分治法為基礎的NGS 序列排序演算法
次世代定序 (NGS) 的技術讓生物學家得以用單一核甘酸的分辨率探討基因體之間的差異,而許多NGS的相關應用都需要能夠快速並準確地將大量的定序片段資料進行序列排比,找出這些片段在基因體中的位置。隨著NGS技術不斷演進,使得定序片段的長度也越來越長,而衍生出新的問題,亦即大部分NGS序列排比方法是針對短序列所開發的,在處理長序列或高錯誤率的資料時有很嚴重的效率落差。此外巨量的短序列片段的排序經常需要大量的計算機運算,因而造成分析上的時間瓶頸。目前主要的NGS短序列排比方法大都基於類似的架構,亦即「種子與延伸」(seed-and-extend)的策略來進行,最典型的例子即是BLAST。種子為序列片段在參考基因體上完全一樣的最長子片段,接著以該種子為中心前後延伸,延伸的過程是耗時最多的步驟,多數方法相異的地方也在於延伸的技巧不同。
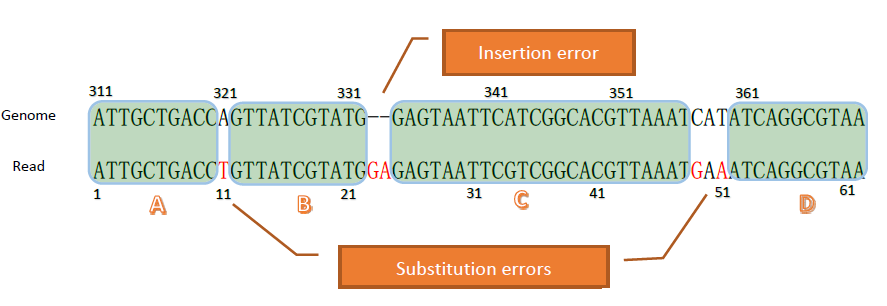
由本核心中央研究院資訊科學所許聞廉老師研究團隊所開發一個快速的短序列排比工具,稱為Kart,利用分治法 (divide-and-conquer)依據短序列在參考基因體上的比對狀況,將短序列切割成不同的區塊,這些區塊代表短序列在參考基因體上完美對應的子序列,而相鄰區塊間通常是很短的缺口片段,主要是基因體變異所造成的缺口,如單核苷酸多態性(SNP)、插入(insertion)或缺失(deletion)變異。由於缺口一般都很短,因此Kart只需要花費少量的計算時間來處理。本方法可大大減少間隔排比的長度,進而降低動態規劃演算法的計算量,這樣的演算法非常適用於相似性高的序列,如次世代定序的每一條短序列都是基因體中某個特定片段所產生的副本,只是當中可能包含某些定序錯誤及變異。由於產生無間隔排比(un-gapped alignment,亦即無插入缺失indels)的速度遠快於有間隔排比, 因此研究團隊將需要排比的次世代定序短序列與參考序列相對應的區域區分為兩類:簡易排比區(simple pair)與一般排比區(normal pair),如圖所示。其中簡易排比區(如圖中的A, B, C及D)具有相同的序列片段,而一般排比區(如圖中因序列差異所產生的缺口)含有相異點,需要無間隔排比或有間隔排比。找出簡易排比區與一般排比區後,這些區域就可以分別並平行地處理,而最終的排比則是這些排比區接合後的結果。我們模擬產生了Illumina和PacBio的定序資料,以及從SRA資料庫與PacBio下載四組真實資料。經過實驗資料驗證,大多數的排比方法在各模擬資料可產生相似的排比結果,其中準確率(accuracy)和召回率(recall rate)都介於97 和99%。從執行時間來看可以發現,Kart是所有比較方法中執行速度最快的演算法。證明Kart在處理NGS序列所花費的時間遠少於目前主流方法。而第三代定序的資料也逐漸增加,現有的次世代短序列排比方法並不能處理第三代定序所產生的長序列,此外第三代定序的長序列有著更多的定序錯誤,特別是Indel錯誤率特別高,這些都大大增加排比的困難度,採用傳統的「種子與延伸」策略將無法有效的處理第三代定序的長序列片段。經過實驗測試,Kart在處理長序列片段也有很好的效率和準確度,我們使用PacBio的超長序列來測試Kart的效能,發現Kart也能在短時間內產生高準確性的排比。
Kart已於2017年中發表於Bioinformatics期刊,Kart原始碼則可於github網站下載。
文獻內容參考:
- Lin,H.-N., and Hsu,W.-L. (2017) Kart: a divide-and-conquer algorithm forNGS read alignment.Bioinformatics,33: 2281–2287.