最新消息
97年9月4-5日「結構生物資訊學研習會」影音內容上線,歡迎觀賞。
網址為:http://www.nhri.org.tw/rr2008/main4.html
最新發表
蛋白質的環形序列重組資料庫 -- CPDB
蛋白質的環形序列重組 (Circular Permutation, CP)可看作是其原本的N與C端被相接,然後在另一處產生開口。自1979首度發現至今,已有許多資料顯示無論是自然或人造的CP,其結構與活性通常有相當高的保留性(conserved),有時伴隨著結構穩定度與功能多樣性上的提升。有鑑於CP已逐漸被應用在許多結構生物學研究或蛋白質工程領域,然目前相關資源稀少且零散,我們發表第一個CP資料庫 (Circular Permutation DataBase, CPDB)。該資料庫採階層式架構,由上自下分別為 Class, fold, cluster 及 pair。此外,CPDB還提供許多工具來協助使用者偵測、分析、比對與檢視蛋白質間的CP關係(請見圖一),並能預測合適的CP切位以減少人為合成時的嘗試錯誤。CPDB 的整合性文獻列表則有利使用者搜尋其感興趣之資訊。此資料庫將為蛋白質折疊與演化之研究提供許多新材料、有助人們發現新穎的蛋白質結構或功能關係,並加速CP於生技產業上的應用。CPDB的網址是http://sarst.life.nthu.edu.tw/cpdb。
Reference:
1. Lo WC, Lee CC, Lee CY, Lyu PC: CPDB: a database of circular permutation in proteins Nucleic Acids Research 2008, doi:10.1093/nar/gkn679 (Database Issue).
2. Lo WC, Lyu PC: CPSARST: an efficient circular permutation search tool applied to the detection of novel protein structural relationships. Genome Biology 2008, 9:R11.
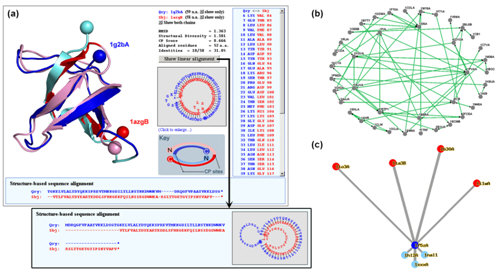 |
圖一:CPDB所提供之多種結構關係檢視工具 |
 |
圖二:CPDB 操作界面總覽。 |
分析工具介紹
CELLO – subCELlular LOcalization predictive system 蛋白質亞細胞位置預測系統
蛋白質為維繫生命最基本的分子物質,在身處的細胞體內的位置內(subcellular localization) 遵行細胞工廠的精密控制,兢兢業業地分工合作著,無論是日常所見的各式動植物,肉眼不可見之微小生物亦然,雖然其調控機制目前並不為科學界全盤了解;一般相信,若能有此線索,可幫助研究者更快速有效的分析該蛋白質的功能;交通大學生物資訊研究團隊(游景盛、陸志豪、陳玉菁等人)所開發的「蛋白質亞細胞位置預測系統」,該研究已發表於蛋白質科學(Proteins Science)和蛋白質(Proteins: Structure, Function, and Bioinformatics)期刊中,以自行研發的獨特演算法發展成分析工具,並建置成網頁伺服器,命名為 CELLO(subCELlular LOcalization predictive system)。使用時僅需提供組成蛋白質之胺基酸序列或是DNA 序列,即可在短時間內自動化、快速且準確的預測該分子可能的細胞位置。我們由序列上根據各種胺基酸特性所萃取的 n-peptide 片段組成份,分析結果優於一般機器學習的演算法和同源性序列辨識法,都可以在許多已知的幾個標準資料組測試上得到極佳的預測結果。在CELLO方法提出後不時有許多相關的研究學者多次來信詢問和表達高度興趣,而 Nature Reviews Microbiology (2006 4:741) 和 Microbiology and Molecular Biology Reviews (2006 70:362)都有詳細的介紹。且相關論文的引用次數 (目前已達 70 次) 亦說明了此工具的高度可信度和推廣的便利性,對於大量 high-throughput 的蛋白質體和基因體資料分析應有相當的幫助。
Reference:
1. Yu CS, Chen YC, Lu CH, Hwang JK. Prediction of protein subcellular localization. Proteins: Sturcture, Function and Bioinformatics (2006), 64, 643-651.
2. Yu CS, Lin CJ, Hwang JK, Predicting Subcellular Localization of Proteins For Gram-Negative Bacteria by Support Vector Machines Based on n-Peptide Compositions. Protein Science (2004), 13, 1402-1406.
3. Gardy, J.L., and F.S.L. Brinkman (2006). "Methods for predicting bacterial protein subcellular localization." Nature Reviews Microbiology 4:741-751.
4. Mee-Jung Han and Sang Yup Lee, The Escherichia coli Proteome: Past, Present, and Future Prospects. Microbiology and Molecular Biology Reviews (2006), 70, 362-439.
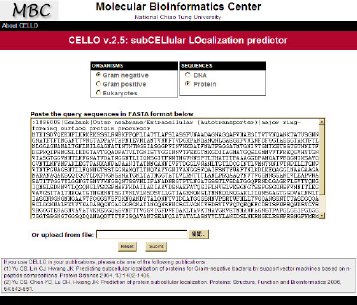 |
圖一:CELLO的操作界面,使用者輸入核酸或蛋白質的序列就能進行分析。 |
探討蛋白質結構「打結」之謎? pKNOT- 環結蛋白質結構資料庫及分析工具
有些蛋白質的直鍵結構會自我交錯產生個結(knot)。這種蛋白質被稱為「環結蛋白質」 (knotted protein)。環結蛋白質是個謎樣的角色 – 蛋白質之環節與其功能有無關聯?環節蛋白質的折疊機制與一般蛋白質的有無差異?在蛋白質合成時,其環節何時發生?蛋白質之環節有多少種?交通大學生物資訊所研究團隊(賴彥龍、顏士中等人)與另一個 Harvard-MIT 團隊在同一期 Nucleic Acids Research 同時發表了第一個環結蛋白質 web server。交通大學團隊的 pKNOT server 提供了現在最齊全的環節蛋白質資料庫。交大團隊發現,在目前PDB中,只存在三種結:3-1結(平結)、4-1結(八字結)、5-2結;總共有七大類的蛋白質家族有節;他們也發現,有些NMR結構被誤解成有節的結構,phytochrome 的八字結被誤認為是平結等等。pKNOT 也提供了環節蛋白質分析工具:使用者上傳蛋白質結構,pKNOT 計算出其簡易平滑結構,使用者可由此輕易的決定其所上傳結構是否有節,或為何種結。pKNOT 能產生蛋白質主鍵平滑過程的 movie 檔案,這除了可幫助使用者,瞭解環結的拓樸特徵,亦可達到對環節蛋白質之教學效果。
Reference:
1. Lai YL, Yen SC, Yu SH, Hwang JK. PKNOT: the Protein KNOT web server, Nucleic Acids Research (2007) 35:W420-W424.
2. Wagner JR, Brunzelle JS, Forest KT, Vierstra RD A light-sensing knot revealed by the structure of the chromophore binding domain of phytochrome. Nature (2005) 438, 325-331.
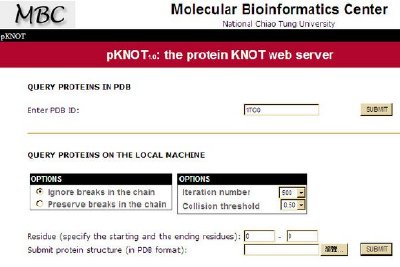 |
圖一: pKNOT server 網站介面,使用者可以輸入蛋白質之 PDB ID 或是上傳自己的 PDB file,就能計算出蛋白質是否具有打結之結構。 |
 |
圖二:為計算出的結果,此為 3-1 環結平結結構。pKNOT 會將結果列出,使用者還能藉由觀看結構影像以了解其打結之位置。如圖,打結處以灰色條帶呈現,使用者可以旋轉或縮放結構影像,以獲得最適當之觀看角度。 |
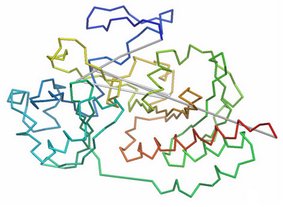
SARST:我把結構比對搜尋的速度加快了
目前蛋白質結構之資訊量已接近指數型成長,因此,人們需要更快速而準確的工具來進行比對搜尋。國立清華大學生物資訊與結構生物研究所呂平江教授實驗室發展了一個高效能的工具:SARST (Structural similarity search Aided by Ramachandran Sequential Transformation)。SARST 透過 nearest-neighbor clustering 的方法將位於 Ramachandran map 上的蛋白質立體結構資訊編碼成純文字字串,並使用繼代式策略 (regenerative approach) 來製作合適的評分表。最後則利用傳統的序列比對搜尋方法來執行結構比對搜尋。SARST 的準確率相近於「組合延展法(CE)」,但在搜尋比對的速度上卻快了將近二十四萬倍。在使用單顆 3.2GHz CPU 之條件下,它能於 0.34 秒內掃描完三萬四千筆蛋白質結構。SARST 有網路服務與單機版本開放大眾使用,網址是 http://sarst.life.nthu.edu.tw/
Reference:
1. Lo WC, Chang CH, Huang PJ, Lyu PC: Protein structural similarity search by Ramachandran codes. BMC Bioinformatics 2007, 8:307.
 |
圖一: SARST 使用介面,使用者可以上傳自己的蛋白質結構 PDB file 來與 PDB 資料庫中所有的結構進行比對,以得到結構相近之蛋白質。 |
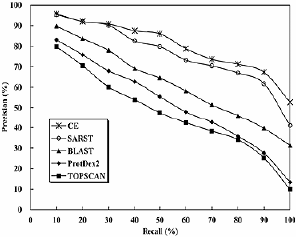 |
圖二: SARST 和世界上其他功能相近之結構比對工具 (包括 CE, BLAST, ProtDex2, TOPSCAN 等),在搜尋結果準確性之比較圖。由比較圖中我們能發現,在這幾種工具當中,SARST 比對之準確性與公認準確度最高的 CE 相去不遠,但速度卻比 CE 快近二十四萬倍。 |
研習會
結構生物資訊學研習會訓練課程介紹
「結構生物資訊學研習會」於9月4-5日在中國醫藥大學互助大樓7F電腦教室舉行。首先由呂平江教授介紹結構生物資訊學的整體概念。呂教授表示,早在1970年代便有生物學家利用電腦來運算序列之結構,當時稱為計算生物學 (Computational Biology)。經過多年發展,現在序列分析工具及資料庫越來越多,使用者必須先想清楚分析之目的以及預期結果,選擇適當的材料做研究,若只是一味的貼上序列,將流於 “Garbage In, Garbage Out” ,不容易得到好的結果。一般而言,當我們得到一條 Genome 序列,第一步可經由 Gene Prediction和Gene Annotation 之分析得知序列中可能存在的 gene 以及 regulatory elements,接著可將核酸序列轉譯成蛋白質序列來做序列比對,得到一個 hypothetical protein。如果我們找到和這個 hypothetical protein 的相似序列,就可以在結構資料庫中尋找這些相似序列的結構,然後藉由homology modeling 的方法來做出結構的預測。而當有了蛋白質結構之後,可以進一步做 molecular docking 或是 virtual screening 之方法,從事藥物的研究及開發。
另一方面,在結構生物學的領域中,藉由軟體來觀察核酸及蛋白質結構的三度空間組成,是很重要的一環,因此呂教授特別在第一天下午介紹 PyMOL 這個蛋白質三度空間結構檢視軟體。課程中藉由軟體操作讓學員觀察蛋白質三級結構,練習將蛋白質結構縮放、旋轉、調整呈現方式、調整顏色、顯示氫鍵、凡得瓦力和模擬表面等基本功能。接著,並介紹蛋白質結構資料庫 PDB (Protein Data Bank) 的使用方法,最後,呂教授另外介紹了由其團隊研發建置的工具-- SARST 和 CPSARST。SARST 是利用 Ramachandran map 蛋白質立體結構資訊快速做結構比對搜尋的工具,詳細說明請參考本期電子報 SARST 專題。CPSARST 則是用來搜尋蛋白質 CP 結購的工具。CP 蛋白質環形序列重組(Circular Permutation) 結構,其性質請參考本期最新發表 CPDB 之介紹。SARST 和 CPSARST 在搜尋比對時不僅快速,還能維持相當高的準確率,是相當便利的兩樣工具。
第二天的研習會介紹結構生物學工具為主。上午首先由黃鎮剛教授介紹CELLO 以及pKNOT二種分析工具。CELLO 是一個 Protein Subcellular Location 的預測工具,在操作介面上,僅需輸入蛋白質序列,系統就會計算出該蛋白質於細胞內主要出現的位置;pKNOT則是預測蛋白質打結的工具,黃教授提到:部分結構在 folding 時會出現「打結」的現象,2005 年 Nature 的一篇論文報導了植物的光敏素 (phytochrome) 蛋白質具有平結之結構,其功能區域即位於打結處,所以蛋白質的打結結構,在了解蛋白質功能上非常值得重視。黃教授實驗室所開發的 pKNOT 蛋白質打結預測工具,可以藉由輸入蛋白質 pdb file,計算出是否有打結的現象發生,並提供打結蛋白之三級結構影像,方便使用者觀察。CELLO 和 pKNOT 之詳細介紹,請見本期相關主題。
第二天下午由楊進木教授介紹了獲得研究發明獎的軟體--GEMDOCK。這個軟體由楊教授實驗室所開發,是以 molecular-ligand docking 的能量計算,來了解當一個 ligand 要和蛋白質結合時,在哪一個位置與構形將最為合適。GEMDOCK 利用蛋白質以及化合物的結構資訊,來預測與蛋白質結合效果最好的化合物,因此是很好的 Structural-based drug design 工具,可用於新藥研發工作。接著楊教授介紹了 fastSCOP 及 3D-partner 二種分析工具,fastSCOP 可以藉由輸入蛋白質結構的 pdb file 與 SCOP 資料庫進行比對,並列出其蛋白質家族的歸屬及其包含的蛋白質 domain ;3D-partner 應用了3D-domain interlogs的理論方法,能讓使用者預測蛋白質間的交互作用。這三種工具對於研究蛋白質交互作用,均能提供很大的幫助。
本次的研討會,學員均表示收穫良多,同時課程的影音節目也已上線,歡迎觀賞,網址為http://www.nhri.org.tw/rr2008/main4.html 。
 |
| 圖一:結構生物資訊學上課情形。 |
分析工具教學
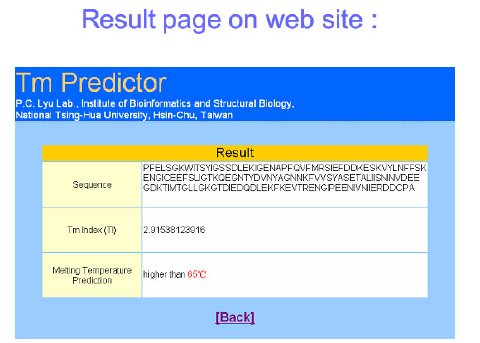
如何使用 TM Predictor (Melting Temperature Prediction Server)
TM Predictor 是由國立清華大學生物資訊與結構生物研究所呂平江教授實驗室建立的,一個預測蛋白質 Tm (melting temperature) 值的工具 。TM Predictor 採用統計方法來分析蛋白質序列與其對熱的耐受度之間的關係。 經由對樣本蛋白 Tm 值與其 dipetide 組成的統計分析,可得一Tm參數表。利用此表預測樣本蛋白的 Tm 值,可達超過 80% 的準確率。我們也應用此方法預測所有已發表之超高溫菌的全基因序列,發現超過72% 的基因產物被判為具高 Tm 值 (Tm > 65 ℃)。這與已發表嗜溫性菌的全基因序列 Tm 值預測結果有顯著的不同。TM Predictor 操作簡單,本文將藉由 4 個步驟說明,教導大家如何使用此一工具。
step1: 連至 TM Predictor 網站
step2: 輸入蛋白質序列,如範例所示的 1a3y 蛋白質

step3: 將序列貼至對話框中 (plain text 格式),並按下 "submit"
step4: 得到分析結果,並預測出蛋白質的 TM 值